Unveiling the Molecular Driving Forces of Pollutant Extraction by Hydrophobic Eutectic Solvents
Pith reviewed 2026-05-08 09:24 UTC · model grok-4.3
The pith
Multiscale simulations show that cooperative hydrogen bonding coupled with dispersion and polarization drives selective pollutant extraction into hydrophobic eutectic solvents.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
The methodology captures the experimentally measured BPA spontaneous migration and thermodynamic stabilization in the HES phase but also identifies the microscopic origin of selectivity: cooperative hydrogen bonding couples to strong dispersion and polarization in the hydrophobic eutectic microenvironment. The robustness of the workflow supports predictive in-silico screening and design of HES formulations for green and sustainable applications.
What carries the argument
The multiscale strategy of monophasic and biphasic molecular dynamics simulations combined with quantum energy decomposition of dominant solvation motifs, which links observable partitioning to the underlying intermolecular forces.
If this is right
- The simulations reproduce the experimental thermodynamic stabilization of BPA in the HES phase.
- Selectivity arises specifically from cooperative hydrogen bonding that is enhanced by dispersion and polarization in the eutectic microenvironment.
- The workflow enables systematic in-silico screening of different HES compositions for targeted pollutant extraction.
- The identified interaction pattern supports rational design of new eutectic formulations instead of empirical testing.
Where Pith is reading between the lines
- The same workflow could be applied to other common pollutants or to eutectic mixtures with different hydrogen-bond donors and acceptors to generate design rules.
- If the force-field accuracy holds across related systems, the approach could shorten the path from molecular insight to industrial-scale green solvent deployment.
- Extending the quantum decomposition to capture temperature or concentration dependence might reveal how the cooperative effects change under operating conditions.
Load-bearing premise
The chosen molecular dynamics force fields and quantum decomposition methods accurately reproduce the real intermolecular interactions and thermodynamic driving forces in hydrophobic eutectic systems without significant systematic errors.
What would settle it
New experimental measurements of BPA partitioning coefficients or solvation energies in the same TOPO-menthol HES that deviate substantially from the simulated values, or higher-level quantum calculations that assign different relative weights to hydrogen bonding versus dispersion and polarization, would indicate the driving forces are not correctly identified.
Figures
read the original abstract
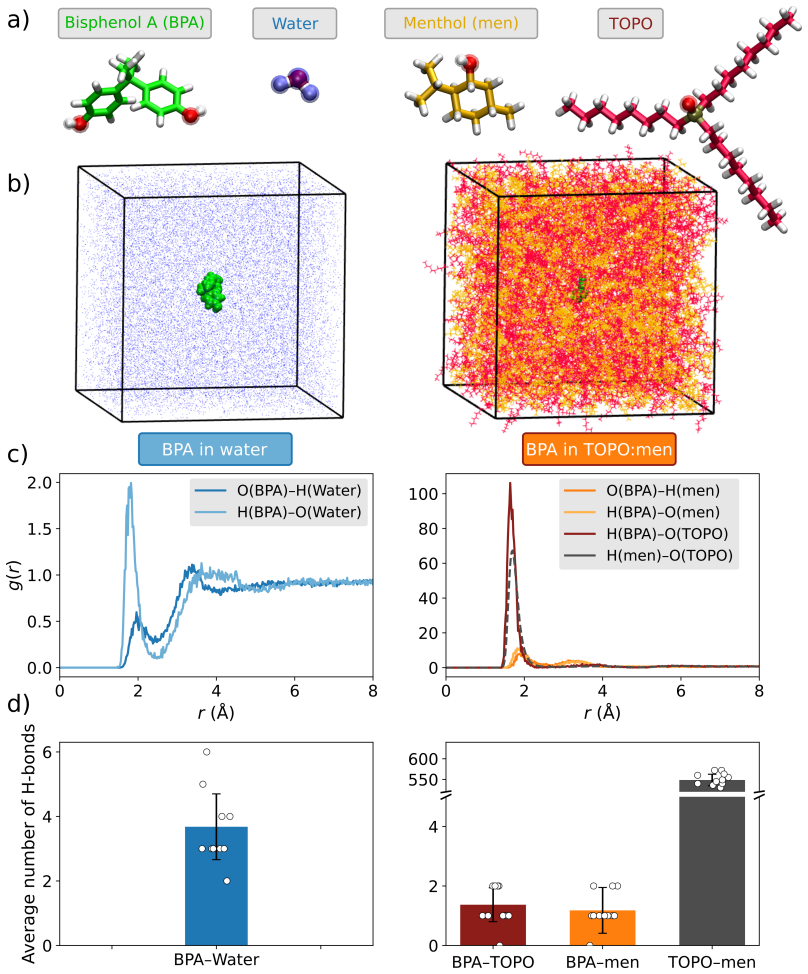
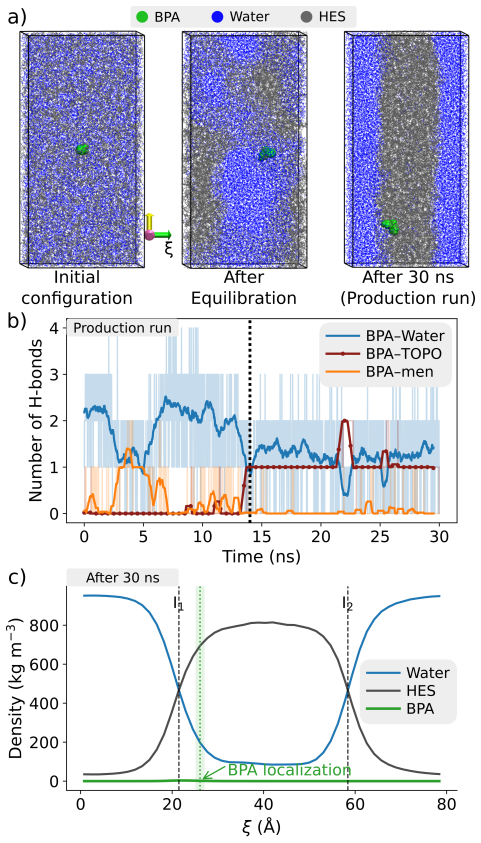
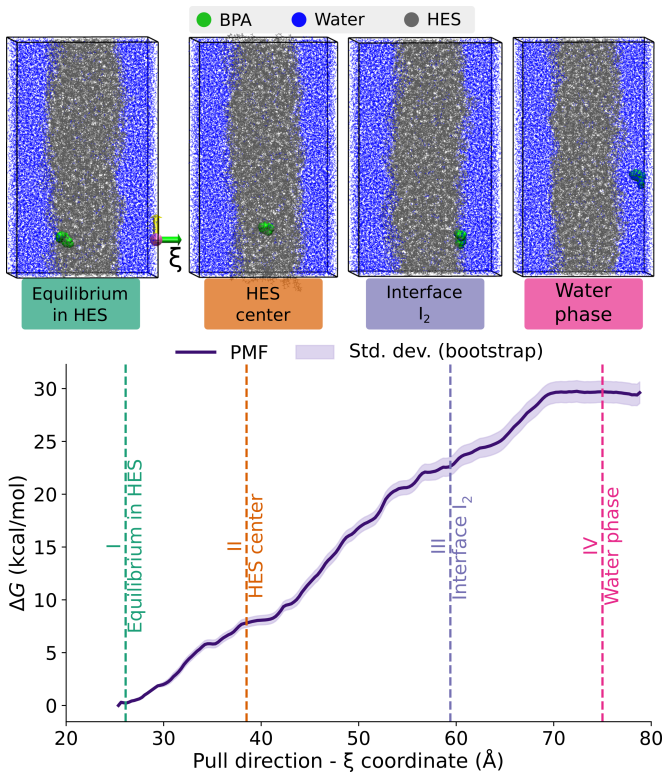
Hydrophobic eutectic solvents (HES) are emerging as sustainable alternatives to conventional organic solvents for the extraction of molecular pollutants from water. Yet, their selectivity remains poorly understood, hindering the predictive design of eutectic solvents beyond empirical success. Here, we present a multiscale strategy to rationalize and predict solute partitioning in HES. Focusing on bisphenol A (BPA) in trioctylphosphine oxide (TOPO):menthol as a prototypical system, we combine monophasic and biphasic molecular dynamics with quantum energy decomposition of dominant solvation motifs. Our methodology captures the experimentally measured BPA spontaneous migration and thermodynamic stabilization in the HES phase but also identifies the microscopic origin of selectivity: cooperative hydrogen bonding couples to strong dispersion and polarization in the hydrophobic eutectic microenvironment. The robustness of our workflow paves the way for the predictive in-silico screening and design of HES formulations for green and sustainable applications.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The paper presents a multiscale computational workflow combining monophasic and biphasic molecular dynamics simulations with quantum-mechanical energy decomposition to study the extraction of bisphenol A (BPA) by the hydrophobic eutectic solvent (HES) TOPO:menthol. It claims that the approach reproduces the experimentally observed spontaneous migration and thermodynamic stabilization of BPA into the HES phase and identifies the microscopic origin of selectivity as cooperative hydrogen bonding coupled to strong dispersion and polarization interactions within the hydrophobic eutectic microenvironment.
Significance. If the quantitative validation and force-field accuracy claims hold, the work would provide a useful framework for rational, in-silico design of HES formulations for sustainable pollutant extraction, moving the field beyond purely empirical screening. The integration of classical MD for partitioning thermodynamics with QM decomposition for interaction analysis is a methodological strength that could be broadly applicable.
major comments (4)
- [Abstract and Results] Abstract and Results sections: The central claim that the methodology 'captures the experimentally measured BPA spontaneous migration and thermodynamic stabilization' is stated without any reported quantitative metrics (e.g., computed transfer free energies, partition coefficients, or direct numerical comparison to experiment). This absence prevents assessment of whether the MD trajectories actually reproduce the sign and magnitude of the experimental driving force.
- [Methods] Methods section: No details are provided on the specific classical force fields (parameters, charges, or references for TOPO, menthol, BPA, and water), simulation lengths, convergence criteria, or any validation against experimental densities, viscosities, or known partition data. Given that fixed-charge fields are known to under-polarize and mis-rank dispersion versus electrostatics in amphiphilic mixtures, this is load-bearing for the reproduction of negative transfer free energy.
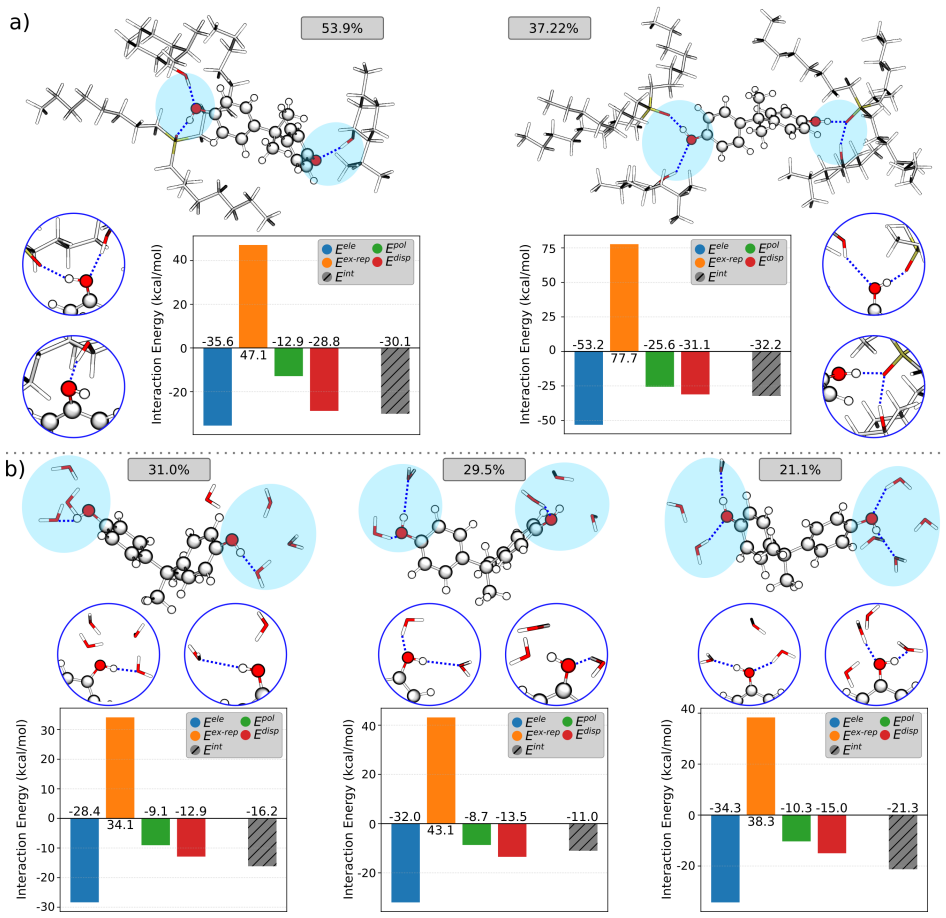
- [Results] Results section on quantum decomposition: The attribution of selectivity to 'cooperative hydrogen bonding couples to strong dispersion and polarization' is presented without numerical values, tables, or error bars from the energy decomposition (e.g., relative magnitudes of H-bond, dispersion, and polarization terms). This leaves the microscopic origin claim qualitative and unverified.
- [Discussion] Discussion: No sensitivity analysis, orthogonal validation (e.g., polarizable force fields or alternative QM schemes), or error propagation is shown to test whether the claimed driving forces survive changes in the underlying model. This is required because the central interpretation collapses if the simulated partition coefficient changes sign under a different but reasonable force field.
minor comments (2)
- [Results] Notation for the HES components and solvation motifs is introduced without a clear table or figure legend summarizing the key species and interaction types.
- [Abstract] The abstract uses 'HES' before the full expansion, which is a minor clarity issue for readers unfamiliar with the acronym.
Simulated Author's Rebuttal
We thank the referee for their thorough and constructive review of our manuscript. The comments have highlighted areas where additional clarity and quantitative detail will strengthen the presentation. We address each major comment below and indicate the revisions we will make.
read point-by-point responses
-
Referee: [Abstract and Results] Abstract and Results sections: The central claim that the methodology 'captures the experimentally measured BPA spontaneous migration and thermodynamic stabilization' is stated without any reported quantitative metrics (e.g., computed transfer free energies, partition coefficients, or direct numerical comparison to experiment). This absence prevents assessment of whether the MD trajectories actually reproduce the sign and magnitude of the experimental driving force.
Authors: We agree that explicit quantitative metrics are needed to fully substantiate the claim of reproducing experimental behavior. The manuscript reports the observation of spontaneous BPA migration into the HES phase in biphasic simulations together with a favorable stabilization relative to the aqueous phase, but does not tabulate computed partition coefficients or transfer free energies with direct numerical comparison to experiment. In the revised manuscript we will add these values, obtained from the existing trajectories via the potential of mean force or counting statistics, and compare them to available experimental partition data for BPA in related hydrophobic solvents. revision: yes
-
Referee: [Methods] Methods section: No details are provided on the specific classical force fields (parameters, charges, or references for TOPO, menthol, BPA, and water), simulation lengths, convergence criteria, or any validation against experimental densities, viscosities, or known partition data. Given that fixed-charge fields are known to under-polarize and mis-rank dispersion versus electrostatics in amphiphilic mixtures, this is load-bearing for the reproduction of negative transfer free energy.
Authors: We apologize for the insufficient detail in the Methods section. The simulations used the OPLS-AA force field with literature-derived parameters and RESP charges for TOPO, menthol, and BPA, together with the TIP3P water model. Production runs were 200 ns following 50 ns equilibration, with convergence assessed by monitoring density, energy, and radial distribution functions. We will expand the Methods section to include all parameter sources, simulation protocols, and any available comparisons to experimental densities and viscosities of the pure HES. This will allow readers to evaluate the force-field choices directly. revision: yes
-
Referee: [Results] Results section on quantum decomposition: The attribution of selectivity to 'cooperative hydrogen bonding couples to strong dispersion and polarization' is presented without numerical values, tables, or error bars from the energy decomposition (e.g., relative magnitudes of H-bond, dispersion, and polarization terms). This leaves the microscopic origin claim qualitative and unverified.
Authors: The quantum energy decomposition analysis was performed on multiple representative solvation clusters extracted from the MD trajectories. While the main text describes the cooperative contributions, the specific numerical magnitudes and uncertainties were placed only in the Supporting Information. We will revise the Results section to include a table of the averaged energy terms (hydrogen bonding, dispersion, polarization) with standard deviations computed across the sampled configurations, thereby rendering the selectivity argument quantitative. revision: yes
-
Referee: [Discussion] Discussion: No sensitivity analysis, orthogonal validation (e.g., polarizable force fields or alternative QM schemes), or error propagation is shown to test whether the claimed driving forces survive changes in the underlying model. This is required because the central interpretation collapses if the simulated partition coefficient changes sign under a different but reasonable force field.
Authors: We recognize that additional robustness checks would be valuable. Performing new simulations with polarizable force fields or alternative QM decomposition schemes lies outside the computational resources available for the present study. In the revised Discussion we will add a dedicated paragraph acknowledging the limitations of fixed-charge models, citing literature on their performance for eutectic solvent mixtures, and reporting error estimates derived from block averaging of the existing data. We maintain that the internal consistency between monophasic and biphasic results supports the reported driving forces, while noting that full sensitivity analysis remains an important direction for follow-up work. revision: partial
Circularity Check
No circularity: standard MD/QM workflow with external experimental comparison
full rationale
The derivation chain relies on monophasic/biphasic MD trajectories using conventional force fields followed by quantum energy decomposition of solvation motifs. These steps are not self-definitional, do not rename fitted parameters as predictions, and do not invoke load-bearing self-citations or uniqueness theorems. The reported agreement with measured BPA migration is a post-simulation validation against independent experiment rather than an input that forces the microscopic interpretation. No equation or workflow step reduces to its own inputs by construction.
Axiom & Free-Parameter Ledger
axioms (2)
- domain assumption Classical molecular dynamics force fields accurately capture solvation, partitioning, and intermolecular interactions in hydrophobic eutectic solvent-water systems
- domain assumption Quantum mechanical energy decomposition reliably quantifies contributions from hydrogen bonding, dispersion, and polarization in dominant solvation motifs
Reference graph
Works this paper leans on
-
[1]
J.; Tu, W.-C.; Levers, O.; Brohl, A.; Hallett, J
(1) Clarke, C. J.; Tu, W.-C.; Levers, O.; Brohl, A.; Hallett, J. P. Green and sustainable solvents in chemical processes.Chem. Rev.2018,118, 747–800. (2) Welton, T. Solvents and sustainable chemistry.Proc. R. Soc. A: Math. Phys. Eng. Sci.2015, 471, 20150502. (3) Schuur, B.; Brouwer, T.; Smink, D.; Sprakel, L. M. Green solvents for sustainable separation p...
work page 2018
-
[2]
(5) Arnold, S. M.; Clark, K. E.; Staples, C. A.; Klecka, G. M.; Dimond, S. S.; Caspers, N.; Hentges, S. G. Relevance of drinking water as a source of human exposure to bisphenol A.J. Expo. Sci. Environ. Epidemiol.2013,23, 137–144. (6) Belfroid, A.; Van Velzen, M.; Van der Horst, B.; Vethaak, D. Occurrence of bisphenol A in surface water and uptake in fish...
work page 2013
-
[3]
(28) Lum, K.; Chandler, D.; Weeks, J
(27) Marcus, Y.Fluctuation theory of solutions: applications in chemistry, chemical engineering, and biophysics; CRC Press Boca Raton, 2013; pp 65–92. (28) Lum, K.; Chandler, D.; Weeks, J. D. Hydrophobicity at small and large length scales
work page 2013
-
[4]
(29) Levy, R. M.; Gallicchio, E. Computer simulations with explicit solvent: recent progress in the thermodynamic decomposition of free energies and in modeling electrostatic effects.Annu. Rev. Phys. Chem.1998,49, 531–567. (30) Szalewicz, K. Symmetry-adapted perturbation theory of intermolecular forces.WIRES: Com- put. Mol. Sci.2012,2, 254–272. (31) Hohen...
work page 1998
-
[5]
(34) Giovannini, T.; G´ omez, S.; Cappelli, C. Modeling Raman Spectra in Complex Environments: From Solutions to Surface-Enhanced Raman Scattering.J. Phys. Chem. Lett.2025,16, 3106–
work page 2025
-
[6]
(35) Pour, S. B.; Sardroodi, J. J.; Ebrahimzadeh, A. R. Structure and dynamics of hydrophobic deep eutectic solvents composed from terpene-fatty acids investigated by molecular dynamics simulation.J. Mol. Graph. Model..2022,114, 108180. (36) Paul, N.; Harish, G.; Banerjee, T. Stability mechanism of menthol and fatty acid based hydrophobic eutectic solvent...
work page 2022
-
[7]
(40) Fan, T.; Yan, Z.; Yang, C.; Qiu, S.; Peng, X.; Zhang, J.; Hu, L.; Chen, L. Preparation of menthol-based hydrophobic deep eutectic solvents for the extraction of triphenylmethane dyes: quantitative properties and extraction mechanism.Analyst2021,146, 1996–2008. (41) Paul, N.; Banerjee, T. Study on the extraction of acetamiprid and imidacloprid from an...
work page 1996
-
[8]
(61) Sousa da Silva, A. W.; Vranken, W. F. ACPYPE-Antechamber python parser interface.BMC research notes2012,5, 1–8. (62) Kagami, L.; Wilter, A.; Diaz, A.; Vranken, W. The ACPYPE web server for small-molecule MD topology generation.Bioinform.2023,39, btad350. (63) Jorgensen, W. L.; Chandrasekhar, J.; Madura, J. D.; Impey, R. W.; Klein, M. L. Comparison of...
work page 2023
-
[9]
(73) G´ omez, S.; Lafiosca, P.; Egidi, F.; Giovannini, T.; Cappelli, C. Uv-resonance Raman spectra of systems in complex environments: A multiscale modeling applied to doxorubicin intercalated into dna.J. Chem. Inf. Model.2023,63, 1208–1217. (74) G´ omez, S.; Lafiosca, P.; Giovannini, T. Modeling UV/Vis absorption spectra of food colorants in solution: An...
work page 2023
-
[10]
(75) Massova, I.; Kollman, P. A. Combined molecular mechanical and continuum solvent approach (MM-PBSA/GBSA) to predict ligand binding.Perspect. Drug Discov. Des.2000,18, 113–
work page 2000
-
[11]
(76) Kollman, P. A.; Massova, I.; Reyes, C.; Kuhn, B.; Huo, S.; Chong, L.; Lee, M.; Lee, T.; Duan, Y.; Wang, W.; others Calculating structures and free energies of complex molecules: combining molecular mechanics and continuum models.Acc. Chem. Res.2000,33, 889–897. (77) Hub, J. S.; De Groot, B. L.; van der Spoel, D. g wham: A Free Weighted Histogram Anal...
work page 2000
-
[12]
(83) Folkestad, S. D. et al. eT 1.0: An open source electronic structure program with emphasis on coupled cluster and multilevel methods.J. Chem. Phys.2020,152, 184103. 32
work page 2020
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.