Structure Prediction and Bonding Analysis of B₁₈Ag₂ Clusters Featuring Double-Ring Motifs
Pith reviewed 2026-05-07 15:41 UTC · model grok-4.3
The pith
B18Ag2 adopts a bent double-ring structure of two stacked B9 rings with axial silver atoms as the global minimum.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
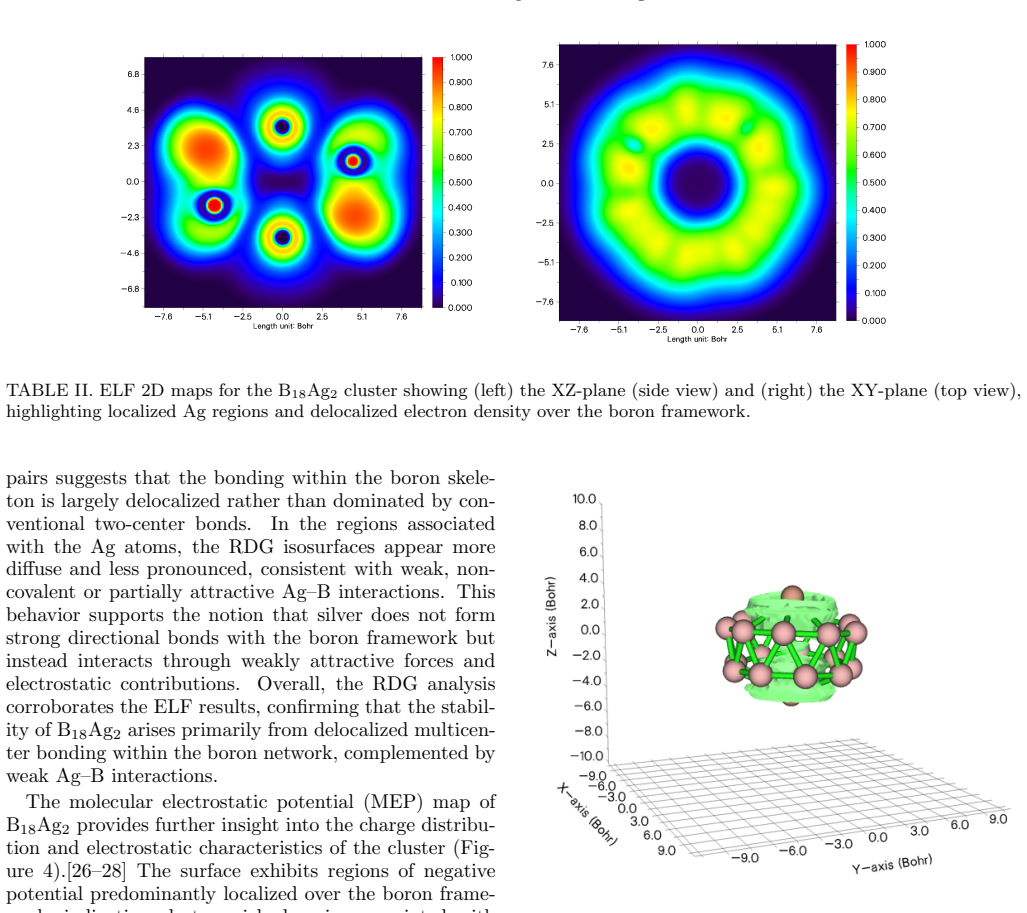
Basin-hopping DFT searches identify a bent double-ring structure as the global minimum for B18Ag2, consisting of two stacked B9 rings symmetrically stabilized by Ag atoms located above and below the boron framework. Real-space analyses using the electron localization function, reduced density gradient, and molecular electrostatic potential establish that bonding is dominated by sigma-delocalization over the boron skeleton, while Ag-B interactions are weak, non-directional, and primarily electrostatic. The continuous annular electron delocalization within the double-ring structure indicates an aromatic-like character. Charge analysis shows moderate electron redistribution from the Ag atoms to
What carries the argument
The bent double-ring motif of two stacked B9 rings symmetrically stabilized by axial Ag atoms.
If this is right
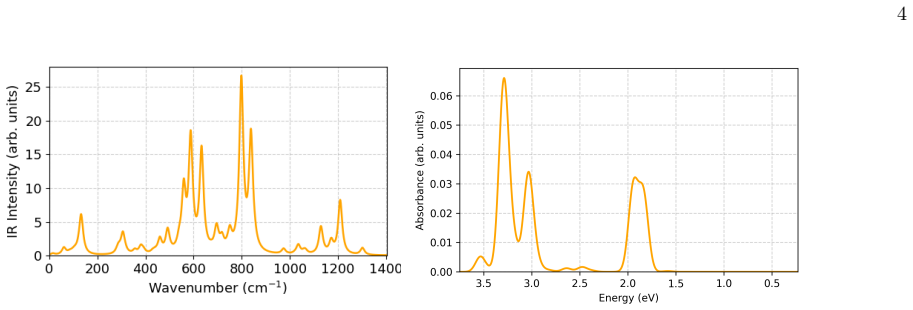
- The UV-Vis absorption spectrum exhibits weak near-infrared transitions and intense bands in the visible and near-ultraviolet regions arising from delocalized excitations in the boron framework.
- Moderate electron density shifts from the Ag atoms to the boron scaffold occur without forming strong directional bonds.
- The cluster possesses aromatic-like character due to continuous annular electron delocalization around the double-ring boron skeleton.
- The Ag atoms function as axial stabilizing centers that modulate the electronic structure while leaving the boron framework as the dominant contributor to stability.
Where Pith is reading between the lines
- The same axial-doping motif could be tested on larger boron rings or tubular boron structures to see whether the stabilization pattern scales.
- Synthesis attempts in the gas phase or on surfaces might succeed under conditions that favor the bent double-ring geometry over planar or cage alternatives.
- Because the silver contribution is mainly electrostatic, replacing Ag with other coinage metals could produce clusters with similar boron-dominated properties but shifted electronic spectra.
Load-bearing premise
The chosen DFT functional and basis set together with the basin-hopping search reliably locate the true global minimum and correctly describe the bonding character without significant method-dependent errors.
What would settle it
A higher-level ab initio calculation or gas-phase spectroscopic measurement that identifies a different isomer as lower in energy or demonstrates directional covalent Ag-B bonding would disprove the reported global minimum and bonding picture.
Figures


read the original abstract
The structural stability, electronic structure, and bonding characteristics of the silver-doped boron cluster B18Ag2 were investigated using density functional theory (DFT) combined with global optimization techniques. Basin-hopping searches identify a bent double-ring structure as the global minimum, consisting of two stacked B9 rings symmetrically stabilized by Ag atoms located above and below the boron framework. The UV-Vis absorption spectrum exhibits weak transitions in the near-infrared region and intense bands in the visible and near-ultraviolet regions, reflecting delocalized electronic excitations within the boron framework. Charge analysis indicates moderate electron redistribution from Ag atoms to the boron scaffold. Real-space bonding analyses based on the electron localization function (ELF), reduced density gradient (RDG), and molecular electrostatic potential (MEP) reveal that bonding is dominated by {$\sigma$}-delocalization over the boron skeleton, while Ag-B interactions are weak, non-directional, and primarily electrostatic. The continuous annular electron delocalization within the double-ring structure suggests an aromatic-like character. These findings establish B18Ag2 as a silver-stabilized boron double-ring cluster in which global electron delocalization governs structural stability, while Ag atoms act as axial stabilizing centers that modulate the electronic structure. This work provides new insight into the role of coinage-metal doping in stabilizing extended boron nanostructures.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript investigates the structure, stability, and bonding of the B18Ag2 cluster using density functional theory combined with basin-hopping global optimization. It identifies a bent double-ring motif consisting of two stacked B9 rings with Ag atoms above and below as the global minimum. The electronic properties, including UV-Vis absorption, charge transfer, and bonding analyzed via ELF, RDG, and MEP, indicate dominant σ-delocalization over the boron skeleton, weak electrostatic Ag-B interactions, and aromatic-like character due to annular electron delocalization.
Significance. Should the structural identification prove robust, the findings advance understanding of how silver doping can stabilize extended boron double-ring structures through axial electrostatic interactions while preserving delocalized boron bonding. The application of multiple real-space analysis tools (ELF, RDG, MEP) provides a detailed bonding picture that could be useful for related cluster systems. The global optimization approach is appropriately chosen for this size of cluster.
major comments (2)
- [Computational Methods] Computational Methods section: The basin-hopping search protocol lacks reported details on the number of independent runs, Monte Carlo steps, temperature schedule, and the specific DFT functional plus basis set used for energy evaluations during the search. This information is load-bearing for the central claim that the bent double-ring is the global minimum, because boron-cluster potential energy surfaces are known to be sensitive to the exchange-correlation approximation and incomplete sampling can miss lower-lying isomers.
- [Results] Results section: No table or figure presents relative energies (or energy differences) of competing isomers such as planar, cage, or differently stacked configurations. Without these comparisons, it is not possible to verify that the reported structure is lower in energy than alternatives by a margin sufficient to support the subsequent bonding analysis.
minor comments (2)
- [Abstract] The abstract and main text use 'aromatic-like character' without a quantitative descriptor (e.g., NICS values or integrated ring-current strength), which would make the claim more precise.
- [Figures] Figure captions for the MEP and RDG plots should explicitly state the isovalue thresholds and color mapping conventions to improve reproducibility.
Simulated Author's Rebuttal
We thank the referee for their positive evaluation of the significance of our findings and for the constructive comments on methodological reporting and results presentation. We address each major comment below and have revised the manuscript accordingly to enhance reproducibility and verifiability.
read point-by-point responses
-
Referee: [Computational Methods] Computational Methods section: The basin-hopping search protocol lacks reported details on the number of independent runs, Monte Carlo steps, temperature schedule, and the specific DFT functional plus basis set used for energy evaluations during the search. This information is load-bearing for the central claim that the bent double-ring is the global minimum, because boron-cluster potential energy surfaces are known to be sensitive to the exchange-correlation approximation and incomplete sampling can miss lower-lying isomers.
Authors: We agree that these details are necessary to substantiate the global optimization results. In the revised manuscript, the Computational Methods section will be expanded to specify the number of independent basin-hopping runs, the number of Monte Carlo steps per run, the temperature schedule, and the DFT functional together with the basis set employed for energy evaluations during the search. This addition will allow assessment of sampling completeness. revision: yes
-
Referee: [Results] Results section: No table or figure presents relative energies (or energy differences) of competing isomers such as planar, cage, or differently stacked configurations. Without these comparisons, it is not possible to verify that the reported structure is lower in energy than alternatives by a margin sufficient to support the subsequent bonding analysis.
Authors: We concur that explicit relative energies are required to confirm the bent double-ring motif as the global minimum. The revised manuscript will incorporate a table (or supplementary figure) listing the relative energies of the reported structure versus other low-lying isomers, including planar, cage-like, and alternative stacking configurations, reported in eV with corresponding symmetries and brief structural descriptors. revision: yes
Circularity Check
No circularity: results from independent numerical optimization and analysis
full rationale
The paper's derivation chain consists of standard DFT computations combined with basin-hopping global searches to locate the lowest-energy isomer of B18Ag2, followed by post-processing analyses (ELF, RDG, MEP, charge distribution, UV-Vis spectra) on the resulting geometry. These steps are not self-referential: the global minimum is identified by exhaustive stochastic search rather than by fitting or definition, and bonding conclusions follow directly from the computed electron density without importing uniqueness theorems or ansatzes from prior self-citations. No equation reduces to its own input by construction, and external literature benchmarks on boron clusters provide independent validation. The approach is self-contained against external methods and data.
Axiom & Free-Parameter Ledger
free parameters (2)
- DFT exchange-correlation functional
- Basis-set size and type
axioms (2)
- standard math Born-Oppenheimer approximation
- domain assumption Basin-hopping locates the true global minimum
Reference graph
Works this paper leans on
- [1]
- [2]
-
[3]
Lopez, Jun Li, and Lai-Sheng Wang
Tian Jian, Wan-Lu Li, Xin Chen, Teng-Teng Chen, Gary V. Lopez, Jun Li, and Lai-Sheng Wang. Com- petition between drum and quasi-planar structures in RhB− 18: motifs for metallo-boronanotubes and metallo- borophenes.Chem. Sci., 7:7020–7027, 2016
work page 2016
-
[4]
Density functional theory investigation on the structure and stability of Sc 2Bn (n=1–10) clusters
Jianfeng Jia, Xiaorong Li, Yanan Li, Lijuan Ma, and Hai-Shun Wu. Density functional theory investigation on the structure and stability of Sc 2Bn (n=1–10) clusters. Computational and Theoretical Chemistry, 1027:128–134, 2014
work page 2014
-
[5]
Wang Zhuan-Yu, Kang Wei-Li, Jia Jian-Feng, and Wu Hai-Shun. Structure and stability of Ti2Bn (n=1-10) clusters: an ab initio investigation.Acta Physica Sinica, 63(23):233102–233102, 2014
work page 2014
-
[6]
Hung Tan Pham, My Phuong Pham-Ho, and Minh Tho Nguyen. Impressive capacity of the B− 7 and V2B7 clusters for CO2 capture.Chemical Physics Letters, 728:186–194, 2019
work page 2019
-
[7]
Hung Tan Pham and Minh Tho Nguyen. Effects of bimetallic doping on small cyclic and tubular boron clus- ters: B 7M2 and B14M2 structures with M = Fe, Co.Phys. Chem. Chem. Phys., 17:17335–17345, 2015
work page 2015
-
[8]
Hai-Ru Li, Ceng Zhang, Wan-Biao Ren, Ying-Jin Wang, and Tao Han. Geometric structures and hydrogen storage properties of M 2B7 (M=Be, Mg, Ca) clusters.Interna- tional Journal of Hydrogen Energy, 48(66):25821–25829, 2023
work page 2023
-
[9]
Rodr´ ıguez-Kessler and Alvaro Mu˜ noz-Castro
P.L. Rodr´ ıguez-Kessler and Alvaro Mu˜ noz-Castro. Struc- ture search for B 7Cr2 clusters: Inverse sandwich struc- ture for the global minimum.Polyhedron, 273:117486, 2025
work page 2025
-
[10]
Bo Le Chen, Wei Guo Sun, Xiao Yu Kuang, Cheng Lu, Xin Xin Xia, Hong Xiao Shi, and George Maroulis. Struc- tural Stability and Evolution of Medium-Sized Tantalum- Doped Boron Clusters: A Half-Sandwich-Structured TaB− 12 Cluster.Inorganic Chemistry, 57(1):343–350, 2018
work page 2018
-
[11]
Cheng Lu, Weiguang Gong, Quan Li, and Changfeng Chen. Elucidating Stress–Strain Relations of ZrB12 from First-Principles Studies.The Journal of Physical Chem- istry Letters, 11(21):9165–9170, 2020
work page 2020
-
[12]
Rodr´ ıguez-Kessler and Alvaro Mu˜ noz-Castro
Peter L. Rodr´ ıguez-Kessler and Alvaro Mu˜ noz-Castro. Al2B7: an extension of the inverse sandwich B 9 cluster featuring Lewis acid sites and planar aromaticity.Phys. Chem. Chem. Phys., 27:12793–12800, 2025
work page 2025
-
[13]
Cabellos-Quiroz, Se- basti´ an Salazar-Colores, P.L
Jose Manuel Guevara-Vela, J.L. Cabellos-Quiroz, Se- basti´ an Salazar-Colores, P.L. Rodr´ ıguez-Kessler, and Al- 6 varo Mu˜ noz-Castro. Structure search for B7Mn2 clusters: Inverse sandwich geometry with a high-spin state.Com- putational and Theoretical Chemistry, 1254:115487, 2025
work page 2025
-
[14]
Rodr´ ıguez-Kessler, Alejandro V´ asquez-Espinal, and Alvaro Mu˜ noz-Castro
P.L. Rodr´ ıguez-Kessler, Alejandro V´ asquez-Espinal, and Alvaro Mu˜ noz-Castro. Structure and stability of Cu- doped Bn (n = 1-12) clusters: DFT calculations.Poly- hedron, 243:116538, 2023
work page 2023
-
[15]
Rodr´ ıguez-Kessler, Alejandro V´ asquez-Espinal, A.R
P.L. Rodr´ ıguez-Kessler, Alejandro V´ asquez-Espinal, A.R. Rodr´ ıguez-Dom´ ınguez, J.L. Cabellos-Quiroz, and A. Mu˜ noz-Castro. Structures of Ni-doped Bn (n = 1-13) clusters: A computational study.Inorganica Chimica Acta, 568:122062, 2024
work page 2024
-
[16]
Probing the weak interaction between silver and boron.Chem
Hyun Wook Choi, Deniz Kahraman, Wei-Jia Chen, and Lai-Sheng Wang. Probing the weak interaction between silver and boron.Chem. Sci., 17:5084–5091, 2026
work page 2026
-
[17]
Alvaro Mu˜ noz-Castro, Ivan A. Popov, and Alexander I. Boldyrev. Long-range magnetic response of toroidal boron structures: B16 and [Co@B16]-/3- species.Phys. Chem. Chem. Phys., 19:26145–26150, 2017
work page 2017
-
[18]
Rodr´ ıguez-Kessler, Salom´ on Rodr´ ıguez-Carrera, and A
David Olalde-L´ opez, P.L. Rodr´ ıguez-Kessler, Salom´ on Rodr´ ıguez-Carrera, and A. Mu˜ noz-Castro. Hydrogen storage properties for bimetallic doped boron clusters M2B7 (M=Fe, Co, Ni).International Journal of Hydro- gen Energy, 2024
work page 2024
-
[19]
The ORCA quantum chemistry program package.The Journal of Chemical Physics, 152(22):224108, 06 2020
Frank Neese, Frank Wennmohs, Ute Becker, and Christoph Riplinger. The ORCA quantum chemistry program package.The Journal of Chemical Physics, 152(22):224108, 06 2020
work page 2020
-
[20]
Carlo Adamo and Vincenzo Barone. Toward reliable den- sity functional methods without adjustable parameters: The PBE0 model.The Journal of Chemical Physics, 110(13):6158–6170, 04 1999
work page 1999
-
[21]
Florian Weigend and Reinhart Ahlrichs. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy.Phys. Chem. Chem. Phys., 7:3297–3305, 2005
work page 2005
-
[22]
Simone Kossmann and Frank Neese. Efficient structure optimization with second-order many-body perturbation theory: The rijcosx-mp2 method.Journal of Chemical Theory and Computation, 6(8):2325–2338, 2010. PMID: 26613489
work page 2010
-
[23]
Tian Lu and Feiwu Chen. Multiwfn: A multifunctional wavefunction analyzer.Journal of Computational Chem- istry, 33(5):580–592, 2012
work page 2012
-
[24]
P. L. Rodr´ ıguez-Kessler, A. R. Rodr´ ıguez-Dom´ ınguez, and Alvaro Mu˜ noz-Castro. Size-dependent structural motifs in Pt nV−(n = 1–13) cluster anions: a DFT in- sight.Journal of Nanoparticle Research, 28(3):59, Feb 2026
work page 2026
-
[25]
Samantha Ortega-Flores, P. L. Rodr´ ıguez-Kessler, and Alvaro Mu˜ noz-Castro. Size-dependent structural motifs in Ag nMo (n = 1-14) clusters: from planar to icosa- hedral architectures.Journal of Nanoparticle Research, 27(10):277, Oct 2025
work page 2025
-
[26]
J. M. Guevara-Vela, P. L. Rodr´ ıguez-Kessler, J. L. Cabellos-Quiroz, T. Rocha-Rinza, A. V´ asquez-Espinal, and A. Mu˜ noz-Castro. Structure- and Size-Dependent Properties of B nCu0/− 2 (n = 2-14) Clusters: DFT Calculations.The Journal of Physical Chemistry A, 129(22):4855–4860, 2025
work page 2025
-
[27]
Peter L. Rodr´ ıguez-Kessler, Ad´ an R. Rodr´ ıguez- Dom´ ınguez, Desmond MacLeod Carey, and Alvaro Mu˜ noz-Castro. Structural characterization, reactivity, and vibrational properties of silver clusters: a new global minimum for ag16.Phys. Chem. Chem. Phys., 22:27255– 27262, 2020
work page 2020
-
[28]
Rodr´ ıguez-Kessler, Alvaro Mu˜ noz-Castro, and Tom´ as Rocha-Rinza
Jos´ e Manuel Guevara-Vela, Peter L. Rodr´ ıguez-Kessler, Alvaro Mu˜ noz-Castro, and Tom´ as Rocha-Rinza. Global minimum structures and electronic stability of Pt-doped silicon clusters PtSin (n = 2 to 11) in neutral and anionic charge states.Phys. Chem. Chem. Phys., pages –, 2026
work page 2026
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.