Recognition: 1 theorem link
· Lean TheoremExpander attention as exchange-correlation
Pith reviewed 2026-05-12 05:16 UTC · model grok-4.3
The pith
An expander graph transformer ansatz yields a linearly scaling non-local exchange-correlation functional that recovers the correct H2 dissociation curve in the strongly correlated regime.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
We propose a linearly scaling non-local XC approximation based on an expander graph transformer ansatz, improving the scaling of O(N squared) or worse for previous ML functionals capable of reliably capturing strongly correlated systems. We show that it recovers the correct H2 dissociation curve in the strongly correlated regime, with promising results on planar H4, a system where even high-level coupled cluster methods break down.
What carries the argument
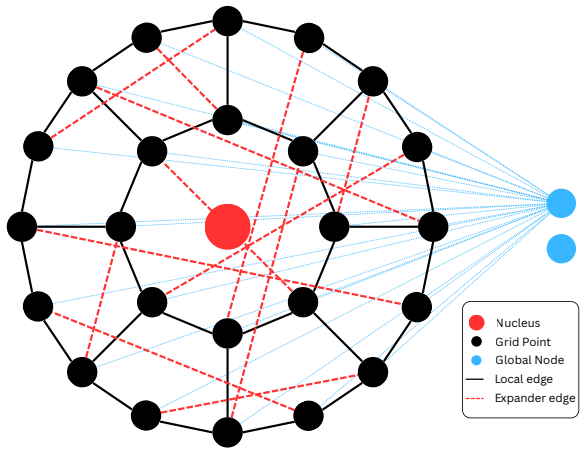
expander graph transformer ansatz for non-local exchange-correlation approximation
If this is right
- Machine-learned functionals become practical for routine calculations on systems too large for previous O(N squared) ML approaches.
- Strongly correlated chemistry such as bond breaking can be treated at DFT cost without sacrificing the correct physical limits.
- The method opens a route to hybrid quantum-classical simulations that combine DFT with more expensive methods only where needed.
- Deployment at scale becomes feasible for materials or biomolecules exhibiting strong correlation.
Where Pith is reading between the lines
- The linear scaling could enable routine treatment of defects or interfaces in materials that current ML functionals cannot reach.
- Similar graph-transformer constructions might transfer to other many-body problems where non-local effects dominate but quadratic scaling is prohibitive.
- Validation on a broader set of molecular benchmarks would be required before claiming transferability beyond the hydrogen examples shown.
Load-bearing premise
The expander graph transformer architecture can faithfully represent true non-local exchange-correlation effects across chemical space without system-specific retraining or uncontrolled errors in the strongly correlated limit.
What would settle it
Application of the functional to a larger hydrogen cluster or another strongly correlated molecule where the computed dissociation or correlation energy deviates significantly from high-accuracy reference values.
Figures




read the original abstract
Kohn-Sham density functional theory (DFT) is the workhorse of quantum chemistry, offering an attractive balance between accuracy and computational cost. Although exact in principle, DFT in practice relies on an approximation to the unknown exchange-correlation (XC) functional, which encodes the many-body quantum effects beyond the mean-field treatment. Many such approximations exist, and machine-learned XC functionals have proliferated in recent years. A persistent challenge in this area is the trade-off between accuracy and computational cost: while high-accuracy ML functionals have shown success on strongly correlated systems that are notoriously difficult for conventional approximations, their unfavorable scaling has limited broader adoption. Here, we propose a linearly scaling non-local XC approximation based on an expander graph transformer ansatz, improving the scaling of $O(N^2)$ or worse for previous ML functionals capable of reliably capturing strongly correlated systems. We show that it recovers the correct $\mathrm{H_2}$ dissociation curve in the strongly correlated regime, with promising results on planar $\mathrm{H_4}$, a system where even high-level coupled cluster methods break down. Our approach thus charts a path toward ML functionals that are both accurate on strongly correlated systems and cheap enough to deploy at scale.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript proposes a machine-learned non-local exchange-correlation functional for Kohn-Sham DFT based on an expander-graph transformer ansatz. It claims this yields linear scaling (improving on O(N^{2}) or worse for prior ML functionals that handle strong correlation), recovers the exact H_{2} dissociation curve in the strongly correlated limit, and gives promising results for planar H_{4}.
Significance. If the linear-scaling claim and numerical results hold after proper validation, the work would address a central practical barrier in DFT by enabling accurate treatment of strongly correlated systems at scale, a longstanding limitation of both conventional and prior ML functionals.
major comments (2)
- [Abstract and Methods] Abstract and Methods: the central claim of linear scaling for the expander-graph transformer XC evaluation is unsupported by any complexity derivation, pseudocode, or empirical timing benchmarks versus system size; without these the asserted improvement over O(N^{2}) priors cannot be assessed and is load-bearing for the paper's contribution.
- [Results] Results section (H_{2}/H_{4} curves): no training details, validation sets, error bars, hyperparameter choices, or comparison baselines are provided for the reported dissociation curves; this leaves open whether the H_{2} recovery and H_{4} results are genuine predictions or lie inside the training distribution, directly affecting the claim of reliable strong-correlation capture.
minor comments (2)
- [Methods] Notation for the expander attention mechanism and its mapping to the XC hole is introduced without a clear equation reference or diagram, making the architecture hard to reproduce from the text alone.
- [Introduction] The manuscript would benefit from explicit citation of recent ML-XC scaling papers to better situate the O(N^{2}) baseline claim.
Simulated Author's Rebuttal
We thank the referee for the thoughtful and constructive report. The comments highlight important areas where the manuscript can be strengthened, particularly regarding explicit support for the scaling claim and transparency in the numerical results. We address each major comment below and will revise the manuscript accordingly.
read point-by-point responses
-
Referee: [Abstract and Methods] Abstract and Methods: the central claim of linear scaling for the expander-graph transformer XC evaluation is unsupported by any complexity derivation, pseudocode, or empirical timing benchmarks versus system size; without these the asserted improvement over O(N^{2}) priors cannot be assessed and is load-bearing for the paper's contribution.
Authors: We agree that the linear-scaling claim requires explicit justification. The expander-graph transformer ansatz uses the bounded degree and expansion properties of expander graphs to restrict attention to a fixed number of neighbors per orbital, yielding O(N) complexity for the non-local XC evaluation. In the revised manuscript we will add a dedicated Methods subsection with a formal complexity derivation, pseudocode for the full XC evaluation pipeline, and empirical wall-time benchmarks on linear hydrogen chains ranging from H_{2} to H_{50}, confirming linear scaling and contrasting it with the O(N^{2}) cost of prior non-local ML functionals. revision: yes
-
Referee: [Results] Results section (H_{2}/H_{4} curves): no training details, validation sets, error bars, hyperparameter choices, or comparison baselines are provided for the reported dissociation curves; this leaves open whether the H_{2} recovery and H_{4} results are genuine predictions or lie inside the training distribution, directly affecting the claim of reliable strong-correlation capture.
Authors: We acknowledge that the absence of these details weakens the interpretability of the numerical results. The revised manuscript will include a new subsection detailing the training protocol: the model was trained on density and energy data from a curated set of small molecules and clusters (with H_{2} and planar H_{4} dissociation curves held out as test cases), the validation split used for hyperparameter selection, error bars obtained from five independent training runs with different random seeds, the chosen hyperparameters (layers, heads, expansion factor, learning rate), and direct comparisons against LDA, PBE, SCAN, and representative prior ML XC functionals. These additions will clarify that the H_{2} recovery occurs on unseen bond lengths in the strongly correlated regime and that the H_{4} results constitute genuine predictions. revision: yes
Circularity Check
No significant circularity; derivation is a proposed ansatz with independent empirical demonstration
full rationale
The manuscript introduces an expander-graph-transformer ansatz as a new non-local XC approximation and reports its performance on H2 dissociation and planar H4. No load-bearing step reduces by construction to a fitted parameter renamed as prediction, nor does any central claim rest on a self-citation chain whose content is unverified. The linear-scaling assertion is presented as a property of the architecture rather than derived from prior self-referential results. Because the work is self-contained against external benchmarks (standard DFT reference curves) and does not smuggle an ansatz via citation or rename a known empirical pattern, the derivation chain does not exhibit circularity.
Axiom & Free-Parameter Ledger
free parameters (1)
- Transformer model weights and hyperparameters
axioms (2)
- standard math Kohn-Sham DFT framework with an approximate XC functional
- domain assumption Expander graphs permit efficient long-range message passing with linear cost
invented entities (1)
-
Expander-attention XC functional
no independent evidence
Reference graph
Works this paper leans on
-
[1]
R. O. Jones. Density functional theory: Its origins, rise to prominence, and future.Reviews of Modern Physics, 87(3):897, 8 2015
work page 2015
-
[2]
Andrea Cavalli, Paolo Carloni, and Maurizio Recanatini. Target-Related Applications of First Principles Quantum Chemical Methods in Drug Design.Chemical Reviews, 106(9):3497–3519, 9 2006
work page 2006
-
[3]
Heriberto Cruz-Martínez, Brenda García-Hilerio, Fernando Montejo-Alvaro, Amado Gazga- Villalobos, Hugo Rojas-Chávez, and Elvia P. Sánchez-Rodríguez. Density Functional Theory- Based Approaches to Improving Hydrogen Storage in Graphene-Based Materials.Molecules 2024, Vol. 29, Page 436, 29(2):436, 1 2024
work page 2024
-
[4]
Masoud Darvish Ganji, Mahyar Rezvani, and Sepideh Tanreh. Characterization and Theoretical Modeling of Solar Cells.Fundamentals of Solar Cell Design, pages 169–215, 1 2023
work page 2023
-
[5]
Lars Stixrude, Nico de Koker, Ni Sun, Mainak Mookherjee, and Bijaya B. Karki. Thermodynam- ics of silicate liquids in the deep Earth.Earth and Planetary Science Letters, 278(3-4):226–232, 2 2009
work page 2009
-
[6]
Inhomogeneous electron gas.Physical review, 136(3B):B864, 1964
Pierre Hohenberg and Walter Kohn. Inhomogeneous electron gas.Physical review, 136(3B):B864, 1964
work page 1964
-
[7]
Norbert Schuch and Frank Verstraete. Computational complexity of interacting electrons and fundamental limitations of density functional theory.Nature Physics 2009 5:10, 5(10):732–735, 8 2009
work page 2009
-
[8]
Susi Lehtola, Conrad Steigemann, Micael J.T. Oliveira, and Miguel A.L. Marques. Recent developments in LIBXC — A comprehensive library of functionals for density functional theory. SoftwareX, 7:1–5, 1 2018
work page 2018
-
[9]
These are the most-cited research papers of all time.Nature, 640(8059):591, 4 2025
Richard Van Noorden. These are the most-cited research papers of all time.Nature, 640(8059):591, 4 2025
work page 2025
-
[10]
Manish Jain, James R. Chelikowsky, and Steven G. Louie. Reliability of Hybrid Functionals in Predicting Band Gaps.Physical Review Letters, 107(21):216806, 11 2011
work page 2011
-
[11]
Spiekermann, Angiras Menon, William H
Xiao Liu, Kevin A. Spiekermann, Angiras Menon, William H. Green, and Martin Head-Gordon. Revisiting a large and diverse data set for barrier heights and reaction energies: best practices in density functional theory calculations for chemical kinetics.Physical Chemistry Chemical Physics, 27(25):13326–13339, 6 2025
work page 2025
- [12]
-
[13]
Stefan Vuckovic, Suhwan Song, John Kozlowski, Eunji Sim, and Kieron Burke. Density Functional Analysis: The Theory of Density-Corrected DFT.Journal of Chemical Theory and Computation, 15(12):6636–6646, 12 2019
work page 2019
-
[14]
A Comprehensive Overview of the DFT-D3 London-Dispersion Correction
Lars Goerigk. A Comprehensive Overview of the DFT-D3 London-Dispersion Correction. Non-Covalent Interactions in Quantum Chemistry and Physics: Theory and Applications, pages 195–219, 1 2017. 10
work page 2017
-
[15]
Li Li, Stephan Hoyer, Ryan Pederson, Ruoxi Sun, Ekin D Cubuk, Patrick Riley, and Kieron Burke. Kohn-Sham equations as regularizer: Building prior knowledge into machine-learned physics.Physical review letters, 126(3):36401, 2021
work page 2021
-
[16]
Muhammad F Kasim and Sam M Vinko. Learning the exchange-correlation functional from na- ture with fully differentiable density functional theory.Physical Review Letters, 127(12):126403, 2021
work page 2021
-
[17]
James Kirkpatrick, Brendan McMorrow, David H.P. Turban, Alexander L. Gaunt, James S. Spencer, Alexander G.D.G. Matthews, Annette Obika, Louis Thiry, Meire Fortunato, David Pfau, Lara Román Castellanos, Stig Petersen, Alexander W.R. Nelson, Pushmeet Kohli, Paula Mori-Sánchez, Demis Hassabis, and Aron J. Cohen. Pushing the frontiers of density functionals b...
work page 2021
-
[18]
Muhammad F Kasim, Susi Lehtola, and Sam M Vinko. DQC: A Python program package for differentiable quantum chemistry.The Journal of chemical physics, 156(8), 2022
work page 2022
-
[19]
Xing Zhang and Garnet Kin Lic Chan. Differentiable quantum chemistry with PySCF for molecules and materials at the mean-field level and beyond.Journal of Chemical Physics, 157(20), 4 2022
work page 2022
-
[20]
Ireneusz W. Bulik, Thomas M. Henderson, and Gustavo E. Scuseria. Can Single-Reference Cou- pled Cluster Theory Describe Static Correlation?Journal of Chemical Theory and Computation, 11(7):3171–3179, 6 2015
work page 2015
-
[21]
Sokolov, Panagiotis Kl Barkoutsos, Pauline J
Igor O. Sokolov, Panagiotis Kl Barkoutsos, Pauline J. Ollitrault, Donny Greenberg, Julia Rice, Marco Pistoia, and Ivano Tavernelli. Quantum orbital-optimized unitary coupled cluster methods in the strongly correlated regime: Can quantum algorithms outperform their classical equivalents?Journal of Chemical Physics, 152(12), 3 2020
work page 2020
-
[22]
Igor O. Sokolov, Gert-Jan Both, Art D. Bochevarov, Pavel A. Dub, Daniel S. Levine, Christo- pher T. Brown, Shaheen Acheche, Panagiotis Kl. Barkoutsos, and Vincent E. Elfving. Quantum- enhanced neural exchange-correlation functionals.Physical Review A, 113(1):012427, 1 2026
work page 2026
-
[23]
Saswata Dasgupta and John M Herbert. Standard grids for high-precision integration of modern density functionals: SG-2 and SG-3.Journal of computational chemistry, 38(12):869–882, 2017
work page 2017
-
[24]
Axel D Becke. A multicenter numerical integration scheme for polyatomic molecules.The Journal of chemical physics, 88(4):2547–2553, 1988
work page 1988
-
[25]
Sutherland, and Ali Kemal Sinop
Hamed Shirzad, Ameya Velingker, Balaji Venkatachalam, Danica J. Sutherland, and Ali Kemal Sinop. Exphormer: Sparse Transformers for Graphs, 7 2023
work page 2023
-
[26]
Expander graphs and their applications
Shlomo Hoory, Nathan Linial, and Avi Wigderson. Expander graphs and their applications. Bulletin of the American Mathematical Society, 43(4):439–561, 10 2006
work page 2006
-
[27]
Alaa El-Din, Ana C C Dutra, and Sam M Vinko
Antonius von Strachwitz, Karim K. Alaa El-Din, Ana C C Dutra, and Sam M Vinko. Data- efficient learning of exchange-correlation functionals with differentiable DFT.Machine Learn- ing: Science and Technology, 7(2):25001, 2 2026
work page 2026
-
[28]
Ryo Nagai, Ryosuke Akashi, and Osamu Sugino. Completing density functional theory by machine learning hidden messages from molecules.npj Computational Materials, 6(1):43, 2020
work page 2020
-
[29]
Benavides-Riveros, and Miguel A.L
Jonathan Schmidt, Carlos L. Benavides-Riveros, and Miguel A.L. Marques. Machine Learning the Physical Nonlocal Exchange–Correlation Functional of Density-Functional Theory.The Journal of Physical Chemistry Letters, 10(20):6425–6431, 10 2019
work page 2019
-
[30]
Yuan Zhuang, Yonghao Gu, Beini Zhang, Jiang Wu, and Guanhua Chen. Machine Learn- ing Accurate Exchange–Correlation Potentials for Reducing Delocalization Error in Density Functional Theory.JACS Au, 5(8):4002–4010, 8 2025. 11
work page 2025
-
[31]
Xiangyun Lei and Andrew J. Medford. Design and analysis of machine learning exchange- correlation functionals via rotationally invariant convolutional descriptors.Physical Review Materials, 3(6):063801, 6 2019
work page 2019
-
[32]
Sebastian Dick and Marivi Fernandez-Serra. Machine learning accurate exchange and correla- tion functionals of the electronic density.Nature Communications 2020 11:1, 11(1):1–10, 7 2020
work page 2020
-
[33]
Adapting hybrid density functionals with machine learning.Sci
Danish Khan, Alastair J A Price, Bing Huang, Maximilian L Ach, and O Anatole V on Lilienfeld. Adapting hybrid density functionals with machine learning.Sci. Adv, 11:31, 2025
work page 2025
-
[34]
Xiaoxun Gong, He Li, Nianlong Zou, Runzhang Xu, Wenhui Duan, and Yong Xu. General framework for E(3)-equivariant neural network representation of density functional theory Hamiltonian.Nature Communications 2023 14:1, 14(1):2848–, 5 2023
work page 2023
-
[35]
Peter Bjørn Jørgensen and Arghya Bhowmik. Equivariant graph neural networks for fast electron density estimation of molecules, liquids, and solids.npj Computational Materials 2022 8:1, 8(1):183–, 8 2022
work page 2022
-
[36]
Nicholas Gao, Eike Eberhard, and Stephan Günnemann. Learning Equivariant Non-Local Electron Density Functionals.13th International Conference on Learning Representations, ICLR 2025, pages 35272–35296, 10 2024
work page 2025
-
[37]
DFTK: A Julian approach for simulating electrons in solids
Michael F Herbst, Antoine Levitt, and Eric Cancès. DFTK: A Julian approach for simulating electrons in solids. InProceedings of the JuliaCon conferences, volume 3, page 69, 2021
work page 2021
-
[38]
Matthias Fey, Jinu Sunil, Akihiro Nitta, Rishi Puri, Manan Shah, Blaž Stojanovi ˇc, Ramona Bendias, Alexandria Barghi, Vid Kocijan, Zecheng Zhang, Xinwei He, Jan Eric Lenssen, and Jure Leskovec. PyG 2.0: Scalable Learning on Real World Graphs.Proceedings of Temporal Graph Learning Workshop, SIGKDD International Conference on Knowledge Discovery and Data M...
work page 2025
-
[39]
V . I. Lebedev. Quadratures on a sphere.USSR Computational Mathematics and Mathematical Physics, 16(2):10–24, 1 1976
work page 1976
-
[40]
Yunsheng Shi, Zhengjie Huang, Shikun Feng, Hui Zhong, Wenjing Wang, and Yu Sun. Masked Label Prediction: Unified Message Passing Model for Semi-Supervised Classification.IJCAI International Joint Conference on Artificial Intelligence, pages 1548–1554, 9 2020
work page 2020
-
[41]
Qiming Sun, Timothy C. Berkelbach, Nick S. Blunt, George H. Booth, Sheng Guo, Zhendong Li, Junzi Liu, James D. McClain, Elvira R. Sayfutyarova, Sandeep Sharma, Sebastian Wouters, and Garnet Kin Lic Chan. PySCF: the Python-based simulations of chemistry framework.Wiley Interdisciplinary Reviews: Computational Molecular Science, 8(1):e1340, 1 2018
work page 2018
-
[42]
SCAN based non-linear double hybrid density functional.Journal of Chemical Physics, 163(14), 10 2025
Danish Khan. SCAN based non-linear double hybrid density functional.Journal of Chemical Physics, 163(14), 10 2025
work page 2025
-
[43]
Thomas N. Kipf and Max Welling. Semi-Supervised Classification with Graph Convolutional Networks.5th International Conference on Learning Representations, ICLR 2017 - Conference Track Proceedings, 9 2016
work page 2017
-
[44]
Justin Gilmer, Samuel S. Schoenholz, Patrick F. Riley, Oriol Vinyals, and George E. Dahl. Neural Message Passing for Quantum Chemistry.34th International Conference on Machine Learning, ICML 2017, 3:2053–2070, 4 2017
work page 2017
-
[45]
Generalized gradient approximation made simple.Physical review letters, 77(18):3865, 1996
John P Perdew, Kieron Burke, and Matthias Ernzerhof. Generalized gradient approximation made simple.Physical review letters, 77(18):3865, 1996
work page 1996
-
[46]
John P Perdew and Yue Wang. Accurate and simple analytic representation of the electron-gas correlation energy.Physical review B, 45(23):13244, 1992
work page 1992
-
[47]
Axel D. Becke. A new mixing of Hartree–Fock and local density-functional theories.The Journal of Chemical Physics, 98(2):1372–1377, 1 1993. 12
work page 1993
-
[48]
Relative expanders or weakly relatively Ramanujan graphs
Joel Friedman. Relative expanders or weakly relatively Ramanujan graphs. https://doi.org/10.1215/S0012-7094-03-11812-8, 118(1):19–35, 5 2003
-
[49]
Diederik P Kingma and Jimmy Lei Ba. Adam: A Method for Stochastic Optimization.3rd Inter- national Conference on Learning Representations, ICLR 2015 - Conference Track Proceedings, 4 2014
work page 2015
-
[50]
Real-space machine learning of correlation density functionals
Elias Polak, Heng Zhao, and Stefan Vuckovic. Real-space machine learning of correlation density functionals. 2024. 13 0.2 0.4 0.6 0.8 1.0 α 0.0 2.5 5.0 7.5 10.0 12.5 /uni27E8d /uni27E9 10 ⟨1 10 0 10 1 α 10 1 10 2 /uni27E8d /uni27E9 Lebedev⟩ l = 11 Lebedev⟩ l = 21 Lebedev⟩ l = 31 Lebedev⟩ l = 41 10 ⋅ α 1.33 Figure 4: Average degree against α parameter for ...
work page 2024
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.