Melting Behavior and Phase Stability of CaO from Neural Network Potentials: a Molecular Dynamics Study
Pith reviewed 2026-06-30 20:20 UTC · model grok-4.3
The pith
The overheating ratio for CaO melting rises with pressure rather than staying fixed.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
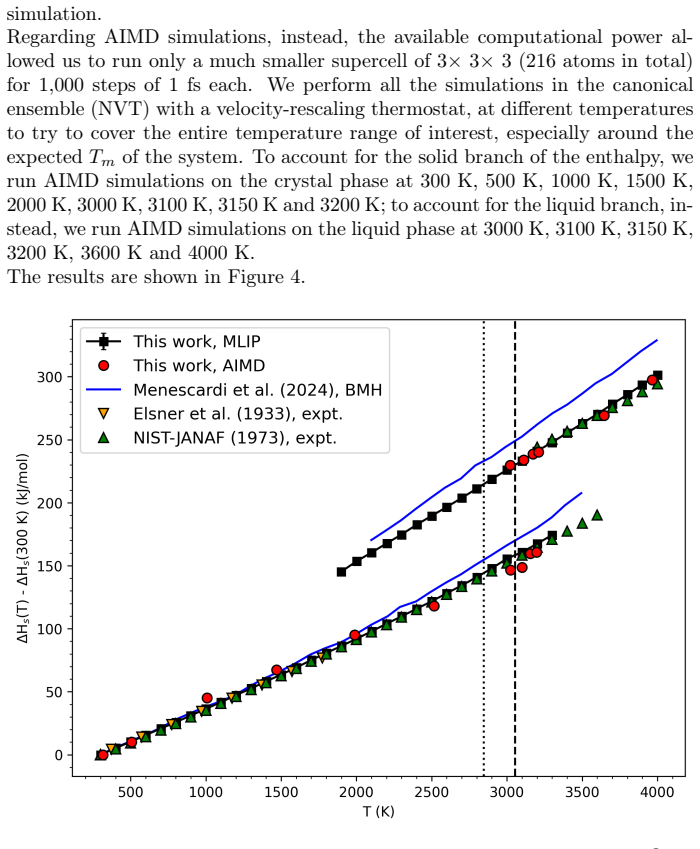
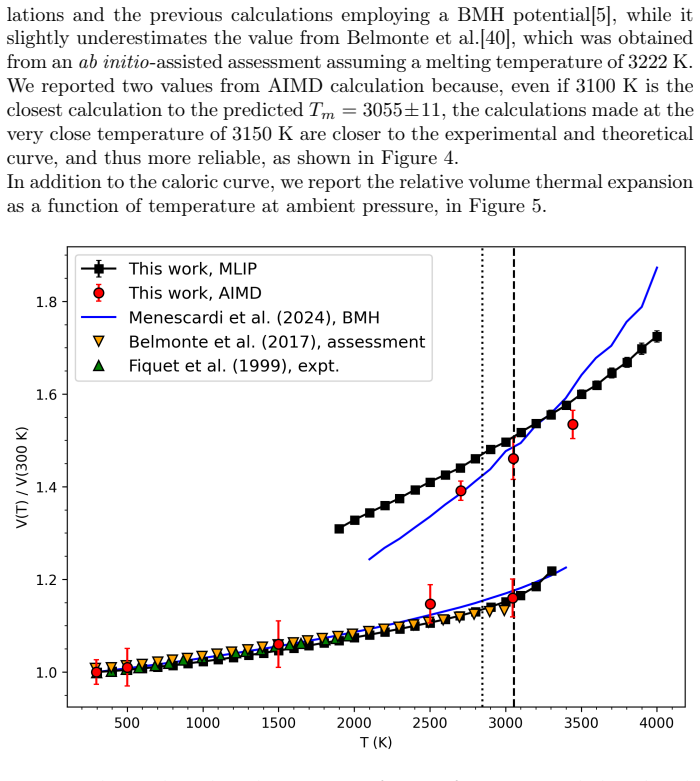
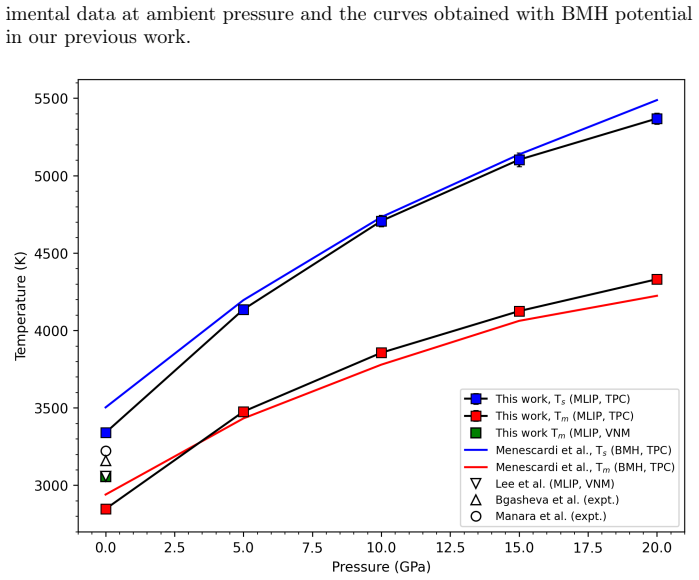
Molecular dynamics simulations driven by a neural network potential trained on PBEsol data give a melting temperature of 2847 K at ambient pressure by the two-phase coexistence method and show an enthalpy of fusion near 73 kJ/mol with a 29 percent volume increase. Extending the same potential to 20 GPa produces a melting curve on which the overheating ratio rises from 17 percent at zero pressure to 24 percent at 20 GPa.
What carries the argument
The neural network interatomic potential trained on solid, liquid, interfacial, and void-containing configurations from PBEsol ab initio molecular dynamics, used inside large-scale molecular dynamics runs with void-nucleated melting and two-phase coexistence protocols.
If this is right
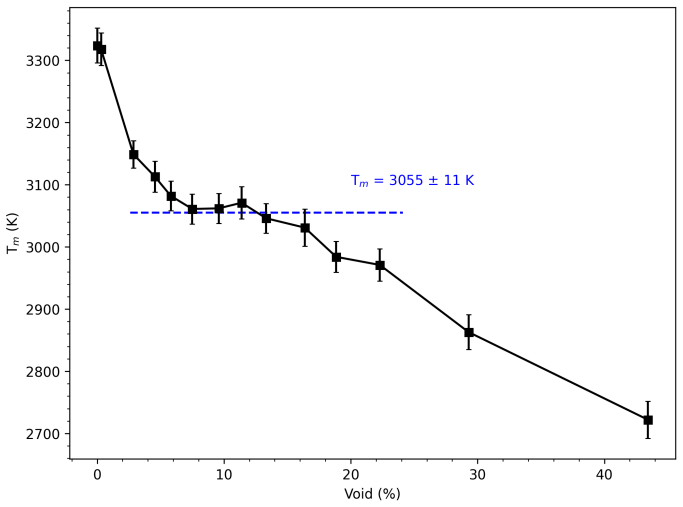
- The ambient-pressure melting temperature is 2847 K by two-phase coexistence and 3055 K by void-nucleated melting.
- The enthalpy of fusion is approximately 73 kJ/mol.
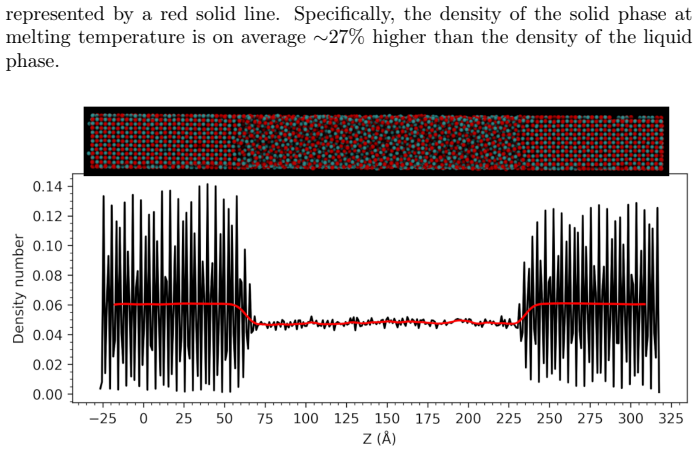
- Density drops by roughly 29 percent on melting at the melting temperature.
- The melting curve is computed up to 20 GPa with one consistent potential.
Where Pith is reading between the lines
- Geophysical models of deep-Earth or planetary mantles that treat the overheating ratio as fixed may need adjustment for pressure dependence.
- The same training and simulation workflow can be reused for other simple ionic oxides where experimental melting data are scarce.
- Direct comparison of the predicted melting curve against future diamond-anvil or shock-wave experiments would test the pressure trend.
Load-bearing premise
The neural network potential reproduces the correct energies and forces for CaO in solid, liquid, and interface states at pressures from zero to 20 GPa.
What would settle it
An experimental melting temperature for CaO at 20 GPa that lies outside the uncertainty range of the simulated melting curve.
Figures
read the original abstract
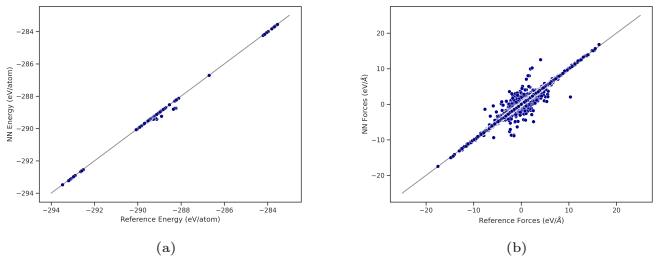
We investigate the melting behavior of calcium oxide (CaO) under extreme conditions, a problem that remains poorly constrained due to experimental limitations despite its relevance for geophysical and technological applications. We develop a Machine Learning Interatomic Potential (MLIP) for CaO with PANNA 2.0 and the LATTE descriptor, training it on a dataset of $\sim$12,000 configurations including solid, liquid, interfacial, and void-containing structures, extracted from ab-initio molecular dynamics data employing PBEsol exchange-correlation functional. We perform large-scale molecular dynamics simulations to compute the melting temperature at ambient pressure using both the void-nucleated melting (VNM) and two-phase coexistence (TPC) methods, obtaining $T_m=3055\pm11$ K and $T_m=2847\pm15$ K, respectively.\\ We calculate an enthalpy of fusion of $\Delta H_f\sim73$ kJ/mol, in agreement with thermodynamic assessments and ab initio calculations. We also reproduce the thermal expansion and obtain a volume increase of $\sim$29% at Tm, consistent with the corresponding decrease in density extracted from spatially resolved number density profiles. Finally, we calculate the high-pressure melting curve of CaO up to 20 GPa, providing one of the very few computational determinations of this quantity to date. The results confirm that the overheating ratio $\eta$ is not constant under pressure, increasing from 17% at ambient pressure to 24% at 20 GPa, confirming previous findings and ruling out the assumption of a fixed overheating ratio. Our results establish MLIP-based simulations as a robust and efficient framework for investigating phase stability in ionic oxides and provide new insight into the melting behavior of CaO under extreme conditions.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The paper trains a PANNA neural network potential on ~12k PBEsol AIMD configurations of CaO (solid, liquid, interfacial, void-containing) and performs large-scale MD to compute melting temperatures via void-nucleated melting (VNM) and two-phase coexistence (TPC) methods, obtaining ambient Tm values of 3055 K and 2847 K. It reports ΔHf ~73 kJ/mol, ~29% volume change on melting, and a high-pressure melting curve to 20 GPa, concluding that the overheating ratio η increases from 17% at ambient pressure to 24% at 20 GPa.
Significance. If the potential is transferable to high pressure, the work supplies one of the few computational melting curves for CaO up to 20 GPa and supplies evidence against a pressure-independent overheating ratio, which is relevant for geophysical modeling of oxides. The use of an independently trained MLIP with no circular fitting to the reported quantities is a strength.
major comments (2)
- [Methods (training dataset)] Methods (training dataset description): the ~12,000 configurations are stated to be extracted from PBEsol AIMD but no information is given on whether the AIMD trajectories sample pressures up to 20 GPa, the corresponding densities, or liquid/interface structures at elevated pressure. This detail is load-bearing for attributing the reported increase in η to physical behavior rather than extrapolation error of the PANNA potential.
- [Results (melting temperatures)] Results (ambient-pressure melting points): the 208 K discrepancy between VNM (3055±11 K) and TPC (2847±15 K) is large enough to affect the baseline value of η; the manuscript must demonstrate which method (or combination) is used for the high-pressure curve and why the difference does not undermine the claimed pressure dependence of η from 17% to 24%.
minor comments (1)
- [Abstract] Abstract and text should explicitly define the overheating ratio η (e.g., (Tm_VNM − Tm_TPC)/Tm_TPC or relative to an external reference) so that the numerical values 17% and 24% can be reproduced from the reported Tm figures.
Simulated Author's Rebuttal
We thank the referee for the constructive comments on our manuscript. We address each major point below and will revise the manuscript to incorporate the requested clarifications and additional details.
read point-by-point responses
-
Referee: Methods (training dataset description): the ~12,000 configurations are stated to be extracted from PBEsol AIMD but no information is given on whether the AIMD trajectories sample pressures up to 20 GPa, the corresponding densities, or liquid/interface structures at elevated pressure. This detail is load-bearing for attributing the reported increase in η to physical behavior rather than extrapolation error of the PANNA potential.
Authors: We agree that the manuscript should explicitly document the pressure and density ranges sampled during AIMD data generation. The training configurations were drawn from AIMD runs performed at multiple pressures up to 25 GPa (including solid, liquid, and interface/void structures at elevated pressure). In the revised manuscript we will add a dedicated paragraph and supplementary table in the Methods section that lists the pressure/density intervals, the number of configurations per interval, and confirms that high-pressure liquid and interface structures are represented in the ~12k set. This addition will allow readers to verify that the reported high-pressure results lie inside the training domain. revision: yes
-
Referee: Results (ambient-pressure melting points): the 208 K discrepancy between VNM (3055±11 K) and TPC (2847±15 K) is large enough to affect the baseline value of η; the manuscript must demonstrate which method (or combination) is used for the high-pressure curve and why the difference does not undermine the claimed pressure dependence of η from 17% to 24%.
Authors: The 208 K offset is expected and arises from the known superheating inherent to the VNM protocol. For the high-pressure melting curve we employed the TPC method to obtain the equilibrium Tm(P) values; the VNM runs were performed only to compute the pressure-dependent overheating ratio η(P) by direct comparison with the TPC baseline at each pressure. We will revise the Results and Discussion sections to state this protocol explicitly, to tabulate both VNM and TPC Tm values at selected high pressures, and to show that the increase in η with pressure remains statistically significant even when the absolute baseline is anchored to the TPC data. A short paragraph will also be added explaining why the method difference does not alter the reported trend in η. revision: yes
Circularity Check
No significant circularity; results computed from MLIP dynamics on independent ab initio training data
full rationale
The derivation trains a PANNA MLIP on ~12k PBEsol AIMD configurations (solid/liquid/interface/void) then runs large-scale MD to obtain VNM and TPC melting points, ΔHf, volume change, and the pressure dependence of η up to 20 GPa. These outputs are generated by the dynamics of the fitted potential rather than being algebraically or statistically forced to equal the training inputs. The paper compares results to external thermodynamic assessments and prior ab initio work; no self-citation is invoked as a uniqueness theorem or load-bearing premise. The training-set pressure range is described only generically, but this affects transferability, not circularity of the reported chain.
Axiom & Free-Parameter Ledger
free parameters (1)
- Neural network weights and biases
axioms (1)
- domain assumption PBEsol exchange-correlation functional provides sufficiently accurate reference data for CaO energetics across solid, liquid, and interface regimes
Reference graph
Works this paper leans on
-
[1]
D. L. Anderson, Theory of the Earth, Blackwell Scientific Publications, 1989
1989
-
[2]
Taylor, Cement Chemistry, Emerald Publishing Limited, 1997
H. Taylor, Cement Chemistry, Emerald Publishing Limited, 1997
1997
-
[3]
S.-M.Liang, R.Schmid-Fetzer, Completethermodynamicdescriptionofthe Mg–Ca–O phase diagram including the Ca–O, Mg–O and CaO–MgO subsystems, Journal of the European Ceramic Society 38 (14) (2018) 4768–4785
2018
-
[4]
Manara, R
D. Manara, R. Böhler, L. Capriotti, A. Quaini, Z. Bao, K. Boboridis, L. Luzzi, A. Janssen, P. Pöml, R. Eloirdi, R. Konings, On the melting be- haviour of calcium monoxide under different atmospheres: A laser heating study, Journal of the European Ceramic Society 34 (6) (2014) 1623–1636. 17
2014
-
[5]
Menescardi, D
F. Menescardi, D. Ceresoli, D. Belmonte, Melting behavior of cao at high temperature and pressure: a molecular dynamics study, The Journal of Physical Chemistry C 128 (43) (2024) 18498–18508
2024
-
[6]
X. Sun, T. Song, Y. Chu, Z. Liu, Z. Zhang, Q. Chen, The high-pressure melting curve of CaO, Solid State Communications 150 (37–38) (2010) 1785–1788
2010
-
[7]
Pellegrini, R
F. Pellegrini, R. Lot, Y. Shaidu, E. Küçükbenli, PANNA 2.0: Efficient neural network interatomic potentials and new architectures, The Journal of Chemical Physics 159 (8) (2023)
2023
-
[8]
F. Pellegrini, S. de Gironcoli, E. Küçükbenli, LATTE: an atomic envi- ronment descriptor based on cartesian tensor contractions, arXiv preprint arXiv:2405.08137 (2024)
-
[9]
R. Lot, F. Pellegrini, Y. Shaidu, E. Küçükbenli, Panna: Properties from artificial neural network architectures, Computer Physics Communications 256 (2020) 107402
2020
-
[10]
J.Behler, M.Parrinello, Generalizedneural-networkrepresentationofhigh- dimensional potential-energy surfaces, Physical Review Letters 98 (14) (2007) 146401
2007
-
[11]
Giannozzi, S
P. Giannozzi, S. Baroni, N. Bonini, M. Calandra, R. Car, C. Cavaz- zoni, D. Ceresoli, G. L. Chiarotti, M. Cococcioni, I. Dabo, A. Dal Corso, S. de Gironcoli, S. Fabris, G. Fratesi, R. Gebauer, U. Gerstmann, C. Gougoussis, A. Kokalj, M. Lazzeri, L. Martin-Samos, N. Marzari, F. Mauri, R. Mazzarello, S. Paolini, A. Pasquarello, L. Paulatto, C. Sbrac- cia, S....
2009
-
[12]
Giannozzi, O
P. Giannozzi, O. Andreussi, T. Brumme, O. Bunau, M. Buon- giorno Nardelli, M. Calandra, R. Car, C. Cavazzoni, D. Ceresoli, M. Cococ- cioni, N. Colonna, I. Carnimeo, A. Dal Corso, S. de Gironcoli, P. Delugas, R. A. DiStasio, A. Ferretti, A. Floris, G. Fratesi, G. Fugallo, R. Gebauer, U. Gerstmann, F. Giustino, T. Gorni, J. Jia, M. Kawamura, H.-Y. Ko, A. Ko...
2017
-
[13]
Giannozzi, O
P. Giannozzi, O. Baseggio, P. Bonfà, D. Brunato, R. Car, I. Carnimeo, C. Cavazzoni, S. de Gironcoli, P. Delugas, F. Ferrari Ruffino, A. Ferretti, N. Marzari, I. Timrov, A. Urru, S. Baroni, Quantum ESPRESSO toward the exascale, The Journal of Chemical Physics 152 (15) (2020) 154105
2020
-
[14]
Troullier, J
N. Troullier, J. L. Martins, Efficient pseudopotentials for plane-wave cal- culations, Physical Review B 43 (3) (1991) 1993–2006. 18
1991
-
[15]
J. P. Perdew, A. Ruzsinszky, G. I. Csonka, O. A. Vydrov, G. E. Scuseria, L. A. Constantin, X. Zhou, K. Burke, Restoring the density-gradient expan- sion for exchange in solids and surfaces, Physical Review Letters 100 (13) (2008) 136406
2008
-
[16]
G. I. Csonka, J. P. Perdew, A. Ruzsinszky, P. H. T. Philipsen, S. Lebègue, J. Paier, O. A. Vydrov, J. G. Ángyán, Assessing the performance of re- cent density functionals for bulk solids, Physical Review B 79 (15) (2009) 155107
2009
-
[17]
Plimpton, Fast parallel algorithms for short-range molecular dynamics, Journal of Computational Physics 117 (1) (1995) 1–19
S. Plimpton, Fast parallel algorithms for short-range molecular dynamics, Journal of Computational Physics 117 (1) (1995) 1–19
1995
-
[18]
A. P. Thompson, H. M. Aktulga, R. Berger, D. S. Bolintineanu, W. M. Brown, P. S. Crozier, P. J. in ’t Veld, A. Kohlmeyer, S. G. Moore, T. D. Nguyen, R. Shan, M. J. Stevens, J. Tranchida, C. Trott, S. J. Plimpton, LAMMPS – a flexible simulation tool for particle-based materials modeling at the atomic, meso, and continuum scales, Computer Physics Communi- c...
2022
-
[19]
W. G. Hoover, Canonical dynamics: Equilibrium phase-space distributions, Physical Review A 31 (3) (1985) 1695–1697
1985
-
[20]
Hong, Melting temperature prediction via first principles and deep learning, Computational Materials Science 214 (2022) 111684
Q.-J. Hong, Melting temperature prediction via first principles and deep learning, Computational Materials Science 214 (2022) 111684
2022
-
[21]
C. M. S. Alvares, G. Deffrennes, A. Pisch, N. Jakse, Thermodynamics and structural properties of CaO: A molecular dynamics simulation study, The Journal of Chemical Physics 152 (8) (2020) 084503
2020
-
[22]
Zhang, E
Y. Zhang, E. J. Maginn, A comparison of methods for melting point cal- culation using molecular dynamics simulations, The Journal of Chemical Physics 136 (14) (2012) 144116
2012
-
[23]
A. B. Belonoshko, N. V. Skorodumova, A. Rosengren, B. Johansson, Melt- ing and critical superheating, Physical Review B 73 (1) (2006) 012201
2006
-
[24]
Z. H. Jin, P. Gumbsch, K. Lu, E. Ma, Melting mechanisms at the limit of superheating, Physical Review Letters 87 (5) (2001) 055703
2001
-
[25]
J. F. Lutsko, D. Wolf, S. R. Phillpot, S. Yip, Molecular-dynamics study of lattice-defect-nucleated melting in metals using an embedded-atom-method potential, Physical Review B 40 (5) (1989) 2841–2855
1989
-
[26]
X. Wang, M. Yang, X. Gai, Y. Sun, B. Cao, J. Chen, M. Liang, F. Tian, L.Li, Acomprehensiveinvestigationontheaccuracyandefficiencyofmeth- ods for melting temperature calculation using molecular dynamics simula- tions, Journal of Molecular Liquids 395 (2024) 123924
2024
-
[27]
Y. Zou, S. Xiang, C. Dai, Investigation on the efficiency and accuracy of methods for calculating melting temperature by molecular dynamics simu- lation, Computational Materials Science 171 (2020) 109156. 19
2020
-
[28]
Bgasheva, T
T. Bgasheva, T. Falyakhov, S. Petukhov, M. Sheindlin, A. Vasin, P. Vervik- ishko, Laser-pulse melting of calcium oxide and some peculiarities of its high-temperature behavior, Journal of the American Ceramic Society 104 (7) (2021) 3461–3477
2021
-
[29]
K. Lee, Y. Park, S. Han, Ab initio construction of full phase diagram of MgO-CaO eutectic system using neural network interatomic potentials, Physical Review Materials 6 (11) (2022) 113802
2022
-
[30]
J. R. Morris, C. Z. Wang, K. M. Ho, C. T. Chan, Melting line of aluminum from simulations of coexisting phases, Physical Review B 49 (5) (1994) 3109–3115
1994
-
[31]
P. M. Agrawal, B. M. Rice, D. L. Thompson, Molecular dynamics study of the melting of nitromethane, The Journal of Chemical Physics 119 (18) (2003) 9617–9627
2003
-
[32]
Q.-J. Hong, A. van de Walle, Solid-liquid coexistence in small systems: A statistical method to calculate melting temperatures, The Journal of Chemical Physics 139 (9) (Sep. 2013)
2013
-
[33]
Wang, X.-W
X.-W. Wang, X.-W. Sun, T. Song, J.-H. Tian, Z.-J. Liu, Prediction of the melting curve and phase diagram for CaO using newly developed inter- atomic potentials, Vacuum 209 (2023) 111717
2023
-
[34]
Asadi, M
E. Asadi, M. A. Zaeem, S. Nouranian, M. I. Baskes, Two-phase solid–liquid coexistence of ni, cu, and al by molecular dynamics simulations using the modified embedded-atom method, Acta Materialia 86 (2015) 169–181
2015
-
[35]
Wang, Z.-J
X. Wang, Z.-J. Liu, J.-S. Feng, M.-R. Chen, L. Li, X.-W. Sun, F. Tian, Construction and application of deep learning potential for cao under high pressure, Computational Materials Science 244 (2024) 113154
2024
-
[36]
Humphrey, A
W. Humphrey, A. Dalke, K. Schulten, VMD: Visual molecular dynamics, Journal of Molecular Graphics 14 (1) (1996) 33–38
1996
-
[37]
Giorgino, Computing 1-d atomic densities in macromolecular simula- tions: The density profile tool for vmd, Computer Physics Communications 185 (1) (2014) 317–322
T. Giorgino, Computing 1-d atomic densities in macromolecular simula- tions: The density profile tool for vmd, Computer Physics Communications 185 (1) (2014) 317–322
2014
-
[38]
Deffrennes, N
G. Deffrennes, N. Jakse, C. M. Alvares, I. Nuta, A. Pasturel, A. Khvan, A. Pisch, Thermodynamic modelling of the Ca–O system including 3rd generation description of cao and CaO2, Calphad 69 (2020) 101764
2020
-
[39]
M, NIST-JANAF Thermochemical Tables, 4th Edition, American Insti- tute of Physics, 1998
C. M, NIST-JANAF Thermochemical Tables, 4th Edition, American Insti- tute of Physics, 1998
1998
-
[40]
Belmonte, G
D. Belmonte, G. Ottonello, M. V. Zuccolini, Ab initio-assisted assessment of the CaO-SiO2 system under pressure, Calphad 59 (2017) 12–30
2017
-
[41]
Gurvich, I
L. Gurvich, I. Veyts, C. Tab Alcock, V. Iorish, Elements b, al, ga, in, tl, be, mg, ca, sr, ba, and their compounds. crc press and begell house. part one. methods and computation. viii+ 707 pp. part two. tables, Thermodynamic Properties of Individual Substances 3 (1994) XV+–448. 20
1994
-
[42]
Fiquet, P
G. Fiquet, P. Richet, G. Montagnac, High-temperature thermal expansion of lime, periclase, corundum and spinel, Physics and Chemistry of Minerals 27 (2) (1999) 103–111
1999
-
[43]
A. S. Arkhipin, A. Pisch, I. A. Uspenskaya, N. Jakse, A molecular dynamics simulation study of crystalline and liquid mgo, Ceramics 7 (3) (2024) 1187– 1203
2024
-
[44]
Zykova-Timan, D
T. Zykova-Timan, D. Ceresoli, U. Tartaglino, E. Tosatti, Physics of solid and liquid alkali halide surfaces near the melting point, The Journal of Chemical Physics 123 (16) (2005) 164701
2005
-
[45]
Gómez, C
L. Gómez, C. Gazza, H. Dacharry, L. Peñaranda, A. Dobry, Pressure de- pendence of the melting mechanism at the limit of overheating in Lennard- Jones crystals, Physical Review B 71 (13) (2005) 134106. 21
2005
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.