MorphoHELM: A Comprehensive Benchmark for Evaluating Representations for Microscopy-Based Morphology Assays
Pith reviewed 2026-05-19 15:42 UTC · model grok-4.3
pith:WDFD5QSB Add to your LaTeX paper
What is a Pith Number?\usepackage{pith}
\pithnumber{WDFD5QSB}
Prints a linked pith:WDFD5QSB badge after your title and writes the identifier into PDF metadata. Compiles on arXiv with no extra files. Learn more
The pith
No existing model outperforms classic computer vision analytic strategies across all settings for microscopy morphology representations.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
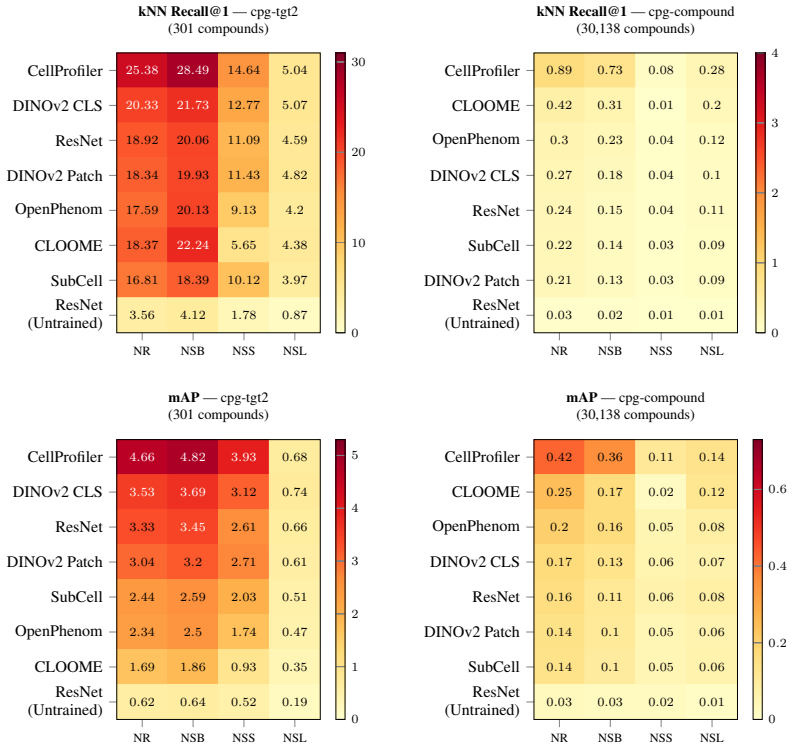
MorphoHELM consolidates tasks and metrics for Cell Painting assays, extends them for robustness, and evaluates the widest range of methods to date while quantifying performance degradation under increasing batch effects; this shows that no model outperforms classic computer vision analytic strategies across all settings, which remain the strongest general use-case representations.
What carries the argument
MorphoHELM benchmark, which applies each representation task at controlled degrees of batch effects to isolate how well methods extract biological signal amid technical noise.
If this is right
- Researchers gain a standardized way to compare new methods against established baselines under realistic noise conditions.
- Method selection should depend on the specific biological signal of interest because of observed performance trade-offs.
- Classic computer vision approaches serve as the default strong choice for general morphological profiling applications.
- Public datasets and code enable consistent future benchmarking and correction of prior fragmented evaluations.
Where Pith is reading between the lines
- The benchmark could be applied to real multi-lab batch variation data to test whether simulated noise levels match actual experimental variability.
- Hybrid methods that combine classic analytic features with deep learning components might close the gap in specific high-noise regimes.
- Similar evaluation structures could help standardize representation testing in other high-content biological imaging domains.
Load-bearing premise
The chosen tasks, metrics, and simulated batch effect levels fully and without bias capture the ability of representations to detect true biological signals.
What would settle it
A new model that outperforms classic computer vision methods on every task at every batch effect level within the MorphoHELM datasets would falsify the central finding.
Figures



read the original abstract
Microscopy images contain rich information about how cells respond to perturbations, making them essential to applications like drug screening. To quantify images, researchers often use representation extraction methods, and recent years have seen a proliferation of deep learning methods. While measuring the quality of these representations is essential, evaluation remains fragmented, with each proposed model evaluated on different tasks and datasets, using custom pipelines and metrics, making it difficult to fairly compare models. Here, we introduce MorphoHELM, a comprehensive open benchmark for evaluating feature extraction methods for Cell Painting, the most widely-used morphological profiling assay. MorphoHELM consolidates evaluation standards in the field, extends and corrects them to be more robust, and evaluates on the widest range of methods to date. A defining feature of the benchmark is that each task is evaluated at different degrees of batch effects (or technical noise), directly quantifying how the ability of methods to detect biological signal degrades as noise increases. Together, these properties enable MorphoHELM to detect trade-offs between methods, and we demonstrate that models that excel at certain kinds of biological signal are weaker at others. We show that no existing model outperforms classic computer vision analytic strategies across all settings, which remain the strongest general use-case representations. All datasets, code, and evaluation tools are publicly available at https://github.com/microsoft/MorphoHELM.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The paper introduces MorphoHELM, a comprehensive open benchmark for evaluating feature extraction methods on Cell Painting microscopy images. It consolidates and extends existing evaluation standards, tests a wide range of methods (deep learning and classic computer vision) at multiple levels of simulated batch effects/technical noise, and concludes that no existing model outperforms classic analytic strategies across all settings, positioning the latter as the strongest general-use representations for detecting biological signals.
Significance. If the central findings hold, the work is significant as a standardization effort in morphological profiling for applications like drug screening. The public release of all datasets, code, and evaluation tools is a clear strength that supports reproducibility and community use. The benchmark's design for quantifying degradation under increasing noise levels usefully reveals method trade-offs, though its impact depends on the fidelity of the noise model to real assay artifacts.
major comments (1)
- [Methods (batch effect simulation and task evaluation)] The batch-effect simulation procedure (described in the methods section on task evaluation under noise) is load-bearing for the headline claim that classic CV strategies remain strongest across settings. The paper does not provide direct validation (e.g., comparison of simulated vs. real multi-batch Cell Painting artifact distributions or spatial correlation statistics) that the chosen noise model (additive Gaussian, global shifts, plate effects) faithfully reproduces the relative difficulty of detecting true morphological perturbations; without this, analytic pipelines tuned to the simulation could appear artificially robust while learned representations are unfairly penalized.
minor comments (2)
- [Results figures] Figure captions and axis labels for the degradation curves under increasing batch effects could be expanded to explicitly state the exact noise parameters and number of replicates per level.
- [Evaluation protocol] The manuscript would benefit from an explicit table listing all evaluated methods, their training regimes (pretrained vs. fine-tuned), and the precise metrics (e.g., mAP, correlation) used for each task.
Simulated Author's Rebuttal
We thank the referee for their constructive comments on our manuscript introducing MorphoHELM. We address the major comment on the batch effect simulation below and outline planned revisions to strengthen the work.
read point-by-point responses
-
Referee: [Methods (batch effect simulation and task evaluation)] The batch-effect simulation procedure (described in the methods section on task evaluation under noise) is load-bearing for the headline claim that classic CV strategies remain strongest across settings. The paper does not provide direct validation (e.g., comparison of simulated vs. real multi-batch Cell Painting artifact distributions or spatial correlation statistics) that the chosen noise model (additive Gaussian, global shifts, plate effects) faithfully reproduces the relative difficulty of detecting true morphological perturbations; without this, analytic pipelines tuned to the simulation could appear artificially robust while learned representations are unfairly penalized.
Authors: We appreciate the referee's emphasis on the need for validation of the batch effect simulation, as it underpins our comparative analysis. The noise models employed—additive Gaussian noise, global shifts, and plate effects—were selected to emulate commonly observed technical artifacts in Cell Painting experiments, drawing from prior literature on batch correction and noise modeling in high-content screening. This controlled simulation enables us to isolate the impact of increasing technical noise on representation quality without confounding factors from real multi-batch data collection. We acknowledge that a direct empirical comparison to real artifact distributions, such as through spatial correlation statistics or distribution matching, is not included in the current manuscript. To address this, we will revise the methods and discussion sections to provide additional justification for the noise model choices with supporting references, explicitly discuss the assumptions and potential limitations of the simulation, and suggest directions for future validation using real multi-batch datasets. This will clarify the scope of our claims regarding method robustness. revision: partial
Circularity Check
No circularity detected in MorphoHELM benchmark
full rationale
The paper introduces MorphoHELM as an open benchmark consolidating evaluation standards for Cell Painting assays on public datasets. It evaluates representation methods across tasks at varying simulated batch effect levels and concludes that classic computer vision analytic strategies remain the strongest general-use representations based on comparative performance. No derivation step reduces to self-definition, fitted inputs renamed as predictions, or load-bearing self-citations; the chain is grounded in external data, metrics, and consolidated standards, making the analysis self-contained against benchmarks.
Axiom & Free-Parameter Ledger
axioms (1)
- domain assumption Standard computer vision and machine learning evaluation practices for image representations apply directly to Cell Painting microscopy data.
Lean theorems connected to this paper
-
IndisputableMonolith/Cost/FunctionalEquation.leanwashburn_uniqueness_aczel unclear?
unclearRelation between the paper passage and the cited Recognition theorem.
We show that no existing model outperforms classic computer vision analytic strategies across all settings, which remain the strongest general use-case representations.
-
IndisputableMonolith/Foundation/Atomicity.leanatomic_tick unclear?
unclearRelation between the paper passage and the cited Recognition theorem.
A defining feature of the benchmark is that each task is evaluated at different degrees of batch effects (or technical noise)
What do these tags mean?
- matches
- The paper's claim is directly supported by a theorem in the formal canon.
- supports
- The theorem supports part of the paper's argument, but the paper may add assumptions or extra steps.
- extends
- The paper goes beyond the formal theorem; the theorem is a base layer rather than the whole result.
- uses
- The paper appears to rely on the theorem as machinery.
- contradicts
- The paper's claim conflicts with a theorem or certificate in the canon.
- unclear
- Pith found a possible connection, but the passage is too broad, indirect, or ambiguous to say the theorem truly supports the claim.
Reference graph
Works this paper leans on
-
[1]
Mark-Anthony Bray, Shantanu Singh, Han Han, Chadwick T Davis, Blake Borgeson, Cathy Hartland, Maria Kost-Alimova, Sigrun M Gustafsdottir, Christopher C Gibson, and Anne E Carpenter. Cell painting, a high-content image-based assay for morphological profiling using multiplexed fluorescent dyes.Nature Protocols, 11(9):1757–1774, August 2016. URL: http: //dx....
-
[2]
Srijit Seal, Maria-Anna Trapotsi, Ola Spjuth, Shantanu Singh, Jordi Carreras-Puigvert, Nigel Greene, Andreas Bender, and Anne E. Carpenter. Cell painting: a decade of discovery and innovation in cellular imaging.Nature Methods, 22(2):254–268, December 2024. URL: http://dx.doi.org/10.1038/s41592-024-02528-8, doi:10.1038/s41592-024-02528-8
-
[3]
Data-analysis strategies for image-based cell profiling
Juan C Caicedo, Sam Cooper, Florian Heigwer, Scott Warchal, Peng Qiu, Csaba Molnar, Aliaksei S Vasilevich, Joseph D Barry, Harmanjit Singh Bansal, Oren Kraus, Mathias Wawer, Lassi Paavolainen, Markus D Herrmann, Mohammad Rohban, Jane Hung, Holger Hennig, John Concannon, Ian Smith, Paul A Clemons, Shantanu Singh, Paul Rees, Peter Horvath, Roger G Linington...
-
[4]
Christian Scheeder, Florian Heigwer, and Michael Boutros. Machine learning and image-based profiling in drug discovery.Current Opinion in Systems Biology, 10:43–52, 2018. URL: http://dx.doi.org/10.1016/j.coisb.2018.05.004, doi:10.1016/j.coisb.2018.05.004
-
[5]
Image-based profiling for drug discovery: due for a machine-learning upgrade?Nat
Srinivas Niranj Chandrasekaran, Hugo Ceulemans, Justin D Boyd, and Anne E Carpenter. Image-based profiling for drug discovery: due for a machine-learning upgrade?Nat. Rev. Drug Discov., 20(2):145–159, February 2021
work page 2021
-
[6]
Anne E Carpenter, Thouis R Jones, Michael R Lamprecht, Colin Clarke, In Han Kang, Ola Friman, David A Guertin, Joo Han Chang, Robert A Lindquist, Jason Moffat, Polina Golland, and David M Sabatini. CellProfiler: image analysis software for identifying and quantifying cell phenotypes.Genome Biol., 7(10):R100, October 2006
work page 2006
-
[7]
David R. Stirling, Madison J. Swain-Bowden, Alice M. Lucas, Anne E. Carpenter, Beth A. Cimini, and Allen Goodman. Cellprofiler 4: improvements in speed, utility and usability. BMC Bioinformatics, 2021. URL: http://dx.doi.org/10.1186/s12859-021-04344-9 , doi:10.1186/s12859-021-04344-9
-
[8]
Evaluating batch correction methods for image-based cell profiling.Nat
John Arevalo, Ellen Su, Jessica D Ewald, Robert van Dijk, Anne E Carpenter, and Shantanu Singh. Evaluating batch correction methods for image-based cell profiling.Nat. Commun., 15(1):6516, August 2024
work page 2024
-
[9]
Qiaosi Tang, Ranjala Ratnayake, Gustavo Seabra, Zhe Jiang, Ruogu Fang, Lina Cui, Yousong Ding, Tamer Kahveci, Jiang Bian, Chenglong Li, Hendrik Luesch, and Yanjun Li. Morphological profiling for drug discovery in the era of deep learning.Briefings in Bioinformatics, 25(4):bbae284, 07 2024. URL: https://doi.org/10.1093/bib/bbae284, arXiv:https://academic.o...
-
[10]
Zitong Chen, Chau Pham, Siqi Wang, Michael Doron, Nikita Moshkov, Bryan A. Plummer, and Juan C Caicedo. CHAMMI: a benchmark for channel-adaptive models in microscopy imaging. InThirty-seventh Conference on Neural Information Processing Systems Datasets and Bench- marks Track. NeurIPS, 2023. URL:https://openreview.net/forum?id=Luc1bZLeMY
work page 2023
-
[11]
Rxrx1: a dataset for evaluating experimental batch correction methods
Maciej Sypetkowski, Morteza Rezanejad, Saber Saberian, Oren Kraus, John Urbanik, James Taylor, Ben Mabey, Mason Victors, Jason Yosinski, Alborz Rezazadeh Sereshkeh, Imran Haque, and Berton Earnshaw. Rxrx1: a dataset for evaluating experimental batch correction methods
- [12]
-
[13]
Learning representations for image-based profiling of perturbations.Nat
Nikita Moshkov, Michael Bornholdt, Santiago Benoit, Matthew Smith, Claire McQuin, Allen Goodman, Rebecca A Senft, Yu Han, Mehrtash Babadi, Peter Horvath, Beth A Cimini, Anne E Carpenter, Shantanu Singh, and Juan C Caicedo. Learning representations for image-based profiling of perturbations.Nat. Commun., 15(1):1594, February 2024. 10
work page 2024
-
[14]
Lazar, Rahul Mohan, Conor Tillinghast, Tommaso Biancalani, Marta M
Safiye Celik, Jan-Christian Hütter, Sandra Melo Carlos, Nathan H. Lazar, Rahul Mohan, Conor Tillinghast, Tommaso Biancalani, Marta M. Fay, Berton A. Earnshaw, and Imran S. Haque. Building, benchmarking, and exploring perturbative maps of transcriptional and morphological data.PLOS Computational Biology, 20(10):1–24, 10 2024. URL: https://doi.org/10. 1371/...
-
[15]
Peter D. Caie, Rebecca E. Walls, Alexandra Ingleston-Orme, Sandeep Daya, Tom Houslay, Rob Eagle, Mark E. Roberts, and Neil O. Carragher. High-content phenotypic profiling of drug response signatures across distinct cancer cells.Molecular Cancer Therapeutics, 9(6):1913–1926, 06 2010. URL: https://doi.org/10.1158/1535-7163.MCT-09-1148 , arXiv:https://aacrjo...
-
[16]
D. Michael Ando, Cory Y . McLean, and Marc Berndl. Improving phe- notypic measurements in high-content imaging screens.bioRxiv, 2017. URL: https://www.biorxiv.org/content/early/2017/07/10/161422, arXiv:https://www.biorxiv.org/content/early/2017/07/10/161422.full.pdf, doi:10.1101/161422
-
[17]
WILDS: A Benchmark of in-the-Wild Distribution Shifts
Pang Wei Koh, Shiori Sagawa, Henrik Marklund, Sang Michael Xie, Marvin Zhang, Akshay Balsubramani, Weihua Hu, Michihiro Yasunaga, Richard Lanas Phillips, Irena Gao, Tony Lee, Etienne David, Ian Stavness, Wei Guo, Berton Earnshaw, Imran Haque, Sara M Beery, Jure Leskovec, Anshul Kundaje, Emma Pierson, Sergey Levine, Chelsea Finn, and Percy Liang. WILDS: A ...
work page 2021
-
[18]
Frank Weber, Guido Knapp, Katja Ickstadt, Günther Kundt, and Änne Glass. Zero- cell corrections in random-effects meta-analyses.Research Synthesis Methods, 11(6):913–919, 2020. URL: https://onlinelibrary.wiley.com/doi/abs/10. 1002/jrsm.1460, arXiv:https://onlinelibrary.wiley.com/doi/pdf/10.1002/jrsm.1460, doi:https://doi.org/10.1002/jrsm.1460
-
[19]
On estimating the relation between blood group and disease.Ann
B Woolf. On estimating the relation between blood group and disease.Ann. Hum. Genet., 19(4):251–253, June 1955
work page 1955
-
[20]
Yemin Yu, Neil Tenenholtz, Lester Mackey, Ying Wei, David Alvarez-Melis, Ava P. Amini, and Alex X. Lu. Causal integration of chemical structures improves representations of microscopy images for morphological profiling. 2025. URL: https://arxiv.org/abs/2504.09544, arXiv:2504.09544
work page internal anchor Pith review Pith/arXiv arXiv 2025
-
[21]
George Tsitsiridis, Ralph Steinkamp, Madalina Giurgiu, Barbara Brauner, Gisela Fobo, Goar Frishman, Corinna Montrone, and Andreas Ruepp. Corum: the comprehensive resource of mam- malian protein complexes–2022.Nucleic Acids Research, 51(D1):D539–D545, 01 2023. URL: https://doi.org/10.1093/nar/gkac1015, arXiv:https://academic.oup.com/nar/article- pdf/51/D1/...
-
[22]
Kevin Drew, John B Wallingford, and Edward M Marcotte. hu.MAP 2.0: integration of over 15,000 proteomic experiments builds a global compendium of human multiprotein assemblies. Mol. Syst. Biol., 17(5):e10016, May 2021
work page 2021
-
[23]
Damian Szklarczyk, Katerina Nastou, Mikaela Koutrouli, Rebecca Kirsch, Farrokh Mehryary, Radja Hachilif, Dewei Hu, Matteo E Peluso, Qingyao Huang, Tao Fang, Nadezhda T Doncheva, Sampo Pyysalo, Peer Bork, Lars J Jensen, and Christian von Mering. The string database in 2025: protein networks with directional- ity of regulation.Nucleic Acids Research, 53(D1)...
-
[24]
Luana Licata, Prisca Lo Surdo, Marta Iannuccelli, Alessandro Palma, Elisa Micarelli, Livia Perfetto, Daniele Peluso, Alberto Calderone, Luisa Castagnoli, and Gianni Ce- sareni. Signor 2.0, the signaling network open resource 2.0: 2019 update.Nucleic Acids Research, 48(D1):D504–D510, 01 2020. URL: https://doi.org/10.1093/nar/ 11 gkz949, arXiv:https://acade...
-
[25]
David Croft, Gavin O’Kelly, Guanming Wu, Robin Haw, Marc Gillespie, Lisa Matthews, Michael Caudy, Phani Garapati, Gopal Gopinath, Bijay Jassal, Steven Jupe, Irina Kalatskaya, Shahana Mahajan, Bruce May, Nelson Ndegwa, Esther Schmidt, Veronica Shamovsky, Christina Yung, Ewan Birney, Henning Hermjakob, Peter D’Eustachio, and Lincoln Stein. Reactome: a datab...
work page 2011
-
[26]
The drug repurposing hub: a next-generation drug library and information resource.Nat
Steven M Corsello, Joshua A Bittker, Zihan Liu, Joshua Gould, Patrick McCarren, Jodi E Hirschman, Stephen E Johnston, Anita Vrcic, Bang Wong, Mariya Khan, Jacob Asiedu, Rajiv Narayan, Christopher C Mader, Aravind Subramanian, and Todd R Golub. The drug repurposing hub: a next-generation drug library and information resource.Nat. Med., 23(4):405–408, April 2017
work page 2017
-
[27]
Kaiming He, Xiangyu Zhang, Shaoqing Ren, and Jian Sun. Deep residual learning for image recognition. In2016 IEEE Conference on Computer Vision and Pattern Recognition (CVPR), volume, 770–778. 2016. doi:10.1109/CVPR.2016.90
-
[28]
Maxime Oquab, Timothée Darcet, Théo Moutakanni, Huy V . V o, Marc Szafraniec, Vasil Khalidov, Pierre Fernandez, Daniel HAZIZA, Francisco Massa, Alaaeldin El-Nouby, Mido Assran, Nicolas Ballas, Wojciech Galuba, Russell Howes, Po-Yao Huang, Shang-Wen Li, Ishan Misra, Michael Rabbat, Vasu Sharma, Gabriel Synnaeve, Hu Xu, Herve Jegou, Julien Mairal, Patrick L...
work page 2024
-
[29]
Masked autoencoders for microscopy are scalable learners of cellular biology
Oren Kraus, Kian Kenyon-Dean, Saber Saberian, Maryam Fallah, Peter McLean, Jess Le- ung, Vasudev Sharma, Ayla Khan, Jia Balakrishnan, Safiye Celik, Dominique Beaini, Maciej Sypetkowski, Chi Vicky Cheng, Kristen Morse, Maureen Makes, Ben Mabey, and Berton Earnshaw. Masked autoencoders for microscopy are scalable learners of cellular biology. In Proceedings...
work page 2024
-
[30]
Ankit Gupta, Zoe Wefers, Konstantin Kahnert, Jan N. Hansen, Mohini K. Misra, Will Leineweber, Anthony Cesnik, Dan Lu, Ulrika Axelsson, Frederic Ballllosera, Russ B. Altman, Theofanis Karaletsos, and Emma Lundberg. Subcell: proteome- aware vision foundation models for microscopy capture single-cell biology.bioRxiv,
-
[31]
URL: https://www.biorxiv.org/content/early/2025/10/30/2024.12.06. 627299, arXiv:https://www.biorxiv.org/content/early/2025/10/30/2024.12.06.627299.full.pdf, doi:10.1101/2024.12.06.627299
-
[32]
CLOOME: contrastive learning unlocks bioimaging databases for queries with chemical struc- tures.Nat
Ana Sanchez-Fernandez, Elisabeth Rumetshofer, Sepp Hochreiter, and Günter Klambauer. CLOOME: contrastive learning unlocks bioimaging databases for queries with chemical struc- tures.Nat. Commun., 14(1):7339, November 2023
work page 2023
-
[33]
Michael Ando, John Arevalo, Melissa Bennion, Nicolas Boisseau, Adriana Borowa, Justin D
Srinivas Niranj Chandrasekaran, Jeanelle Ackerman, Eric Alix, D. Michael Ando, John Arevalo, Melissa Bennion, Nicolas Boisseau, Adriana Borowa, Justin D. Boyd, Laurent Brino, Patrick J. Byrne, Hugo Ceulemans, Carolyn Ch’ng, Beth A. Cimini, Djork-Arne Clevert, Nicole Deflaux, John G Doench, Thierry Dorval, Regis Doyonnas, Vincenza Dragone, Ola Engkvist, Pa...
-
[34]
URL: https://www.biorxiv.org/content/early/2023/03/27/2023.03.23. 534023, arXiv:https://www.biorxiv.org/content/early/2023/03/27/2023.03.23.534023.full.pdf, doi:10.1101/2023.03.23.534023
-
[35]
Mark-Anthony Bray, Sigrun M Gustafsdottir, Mohammad H Rohban, Shantanu Singh, Veb- jorn Ljosa, Katherine L Sokolnicki, Joshua A Bittker, Nicole E Bodycombe, Vlado Dan ˇcík, Thomas P Hasaka, Cindy S Hon, Melissa M Kemp, Kejie Li, Deepika Walpita, Mathias J Wawer, Todd R Golub, Stuart L Schreiber, Paul A Clemons, Alykhan F Shamji, and Anne E Carpenter. A da...
-
[36]
Not Same Batch (NSB): Candidates from the same experimental batch are excluded, testing robustness to day-to-day variation between batches
-
[37]
Not Same Source (NSS): Candidates from the same institution are excluded, testing robust- ness to domain shifts such as microscopy hardware differences
-
[38]
Not Same Layout (NSL): Candidates sharing the same plate position as the query are excluded, controlling for position-induced batch effects [5]. For the enrichment tasks (MoA and gene pathway enrichment), stringency is implemented by constructing separate consensus profiles from non-overlapping subsets of replicates. At the No Restriction level, all repli...
-
[39]
This step both standardizes dimensionality and denoises the feature space
Platewise Center Scaling and PCA.All features are scaled to a standard range, then reduced via Principal Component Analysis applied across the entire dataset. This step both standardizes dimensionality and denoises the feature space. We use 64 components for the PCA analysis
-
[40]
Platewise Robust Standardization.A Median Absolute Deviation (MAD) "Robustize" transformation is applied per plate to align distributions across experimental runs while remaining robust to outliers. This pipeline is applied identically to all methods evaluated in the benchmark. 20 Appendix D: Supplementary Analyses D.1 Effect of Modified Haldane-Anscombe ...
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.