UniField: RBF-Guided Electron Density Fusion for Enhanced Molecular Representations
Pith reviewed 2026-06-30 17:40 UTC · model grok-4.3
The pith
Fusing continuous electron density fields with discrete molecular graphs via RBF-guided equivariant fusion produces new state-of-the-art accuracy on quantum property benchmarks.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
UniField establishes that an SE(3)-equivariant multimodal architecture can intrinsically intertwine discrete topological graphs with continuous quantum electronic environments through RBF-guided fusion of ED point clouds, producing new state-of-the-art results across the ED5-OE, QM9-ED, and QMugs-ED benchmarks with measured gains of 14.8 percent over topology-only SOTA, 37.0 percent over pure-ED models, and 28.2 percent average precision on frontier orbitals.
What carries the argument
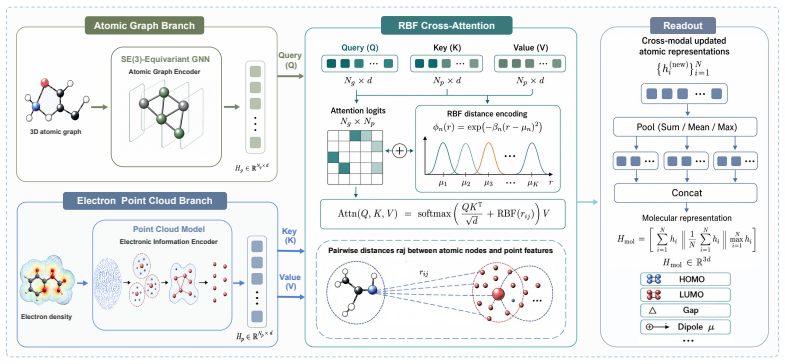
RBF-Guided Electron Density Fusion, the alignment and integration step that combines high-fidelity continuous ED point clouds with discrete graphs inside the SE(3)-equivariant network.
If this is right
- Topology-only models encounter a performance ceiling on tasks involving long-range delocalization that multimodal ED fusion can exceed.
- Predictions for complex drug-like molecules on QMugs-ED become more accurate, especially for frontier orbital properties.
- A standardized, natively aligned benchmark now exists for testing future electron-density-enhanced molecular models.
- Next-generation computational chemistry tools can incorporate continuous quantum fields without abandoning graph-based representations.
Where Pith is reading between the lines
- The same fusion approach could be tested on experimental rather than computed density data to check whether gains persist outside simulated benchmarks.
- Scaling the point-cloud representation to very large systems might reveal memory or alignment bottlenecks not visible in the current datasets.
- Neighboring problems such as protein-ligand binding or materials property prediction could benefit from analogous continuous-field fusion if aligned data can be generated.
Load-bearing premise
The benchmark's ED point clouds accurately represent the continuous electron density field and remain properly aligned with the corresponding graphs so that fusion captures real physical effects rather than artifacts.
What would settle it
Training and evaluating a version of UniField on the same benchmarks but with the ED point clouds randomly permuted or replaced by noise, then observing that predictive performance drops to or below the level of a topology-only baseline, would falsify the value of the fusion.
Figures
read the original abstract
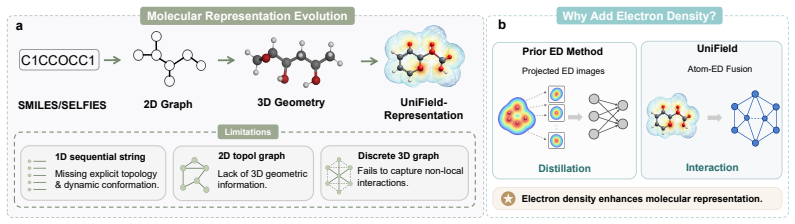
Current 3D geometric molecular representations predominantly focus on discrete atomic skeletons, inherently overlooking the continuous electron density (ED) field that fundamentally governs microscopic quantum behaviors. Consequently, these purely topological models suffer from critical representational blind spots, particularly in capturing long-range electron delocalization and non-covalent interactions, imposing a severe theoretical ceiling on predicting complex quantum properties. To bridge this physical gap and standardize research in electron density-enhanced molecular learning, we first construct the large-scale UniField-ED Benchmark. Comprising the QM9-ED and QMugs-ED datasets, this benchmark provides natively aligned discrete graphs and high-fidelity ED point clouds. Building upon this data infrastructure, we introduce UniField, an SE(3)-equivariant multimodal architecture that intrinsically intertwines discrete topological graphs with continuous quantum electronic environments. Extensive empirical evaluations across all three benchmarks demonstrate that UniField establishes new state-of-the-art performance. Specifically, UniField achieves a 14.8% improvement in overall predictive performance against the leading topology-only SOTA on the ED5-OE benchmark, alongside a 37.0% performance gain over top pure-ED models. Furthermore, on the complex drug-like dataset QMugs-ED, it yields a striking 28.2% average precision improvement across frontier orbital properties. Alongside new SOTA results on QM9-ED, our method establishes a rigorous foundation for next-generation computational chemistry. Code and datasets are anonymously available at https://anonymous.4open.science/r/UniField-ED-5B1B.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript constructs the UniField-ED benchmark (QM9-ED and QMugs-ED) supplying natively aligned discrete molecular graphs and high-fidelity electron density point clouds. It proposes UniField, an SE(3)-equivariant multimodal model that fuses these graphs with continuous ED fields via RBF guidance, and reports new state-of-the-art results: 14.8% overall improvement versus the leading topology-only model on ED5-OE, 37% gain over top pure-ED models, and 28.2% average precision improvement on frontier orbital properties for the drug-like QMugs-ED set, plus new SOTA on QM9-ED.
Significance. If the ED point clouds prove to be high-fidelity samplings of the true continuous field and remain natively aligned with the graphs, the work could meaningfully raise the representational ceiling for properties governed by long-range delocalization and non-covalent interactions. The public release of code and datasets would further strengthen its utility for the field.
major comments (2)
- [Abstract] Abstract: the central SOTA claims (14.8% on ED5-OE versus topology SOTA; 28.2% on QMugs-ED frontier orbitals) rest on the premise that the supplied ED point clouds are both high-fidelity representations of the continuous quantum field and natively aligned to the discrete graphs. No quantitative validation (integrated density error versus grid DFT references, alignment error statistics, or sampling-density sensitivity) is supplied to rule out discretization artifacts or data leakage as alternative explanations for the reported gains.
- [Abstract] The manuscript provides no architecture diagram, loss formulation, or training protocol for the RBF-guided fusion mechanism, making it impossible to assess whether the multimodal gains are attributable to genuine ED incorporation or simply to increased model capacity.
minor comments (1)
- [Abstract] The title references RBF guidance, yet the abstract does not specify how radial basis functions are used to interpolate or condition the continuous ED field onto the discrete graph.
Simulated Author's Rebuttal
We thank the referee for the constructive feedback. We address the two major comments point by point below. Where the comments correctly identify gaps, we commit to revisions that will incorporate the requested elements.
read point-by-point responses
-
Referee: [Abstract] Abstract: the central SOTA claims (14.8% on ED5-OE versus topology SOTA; 28.2% on QMugs-ED frontier orbitals) rest on the premise that the supplied ED point clouds are both high-fidelity representations of the continuous quantum field and natively aligned to the discrete graphs. No quantitative validation (integrated density error versus grid DFT references, alignment error statistics, or sampling-density sensitivity) is supplied to rule out discretization artifacts or data leakage as alternative explanations for the reported gains.
Authors: We agree that quantitative validation of the ED point clouds is necessary to support the SOTA claims and exclude artifacts or leakage. In the revised manuscript we will add a dedicated subsection (Methods) reporting integrated density errors versus grid DFT references, alignment error statistics between graphs and point clouds, and sampling-density sensitivity ablations. These results will also appear in the supplement. revision: yes
-
Referee: [Abstract] The manuscript provides no architecture diagram, loss formulation, or training protocol for the RBF-guided fusion mechanism, making it impossible to assess whether the multimodal gains are attributable to genuine ED incorporation or simply to increased model capacity.
Authors: The full manuscript contains an architecture diagram (Figure 1), the RBF-guided fusion loss (Equation 4, Section 3.2), and the complete training protocol (Section 4.1). To ensure these elements are immediately visible and to directly address capacity concerns, we will insert a short overview paragraph describing the fusion mechanism into the abstract and add an explicit capacity-matched ablation discussion in the main text. revision: partial
Circularity Check
No circularity; empirical benchmark results are independent of any self-referential derivation
full rationale
The paper constructs new datasets (QM9-ED, QMugs-ED) and reports empirical performance gains of a multimodal model on those datasets. No equations, uniqueness theorems, or fitted parameters are presented that reduce a claimed prediction back to the input by construction. The central claims rest on benchmark numbers rather than any derivation chain, and the provided abstract and description contain no self-citation load-bearing steps or ansatz smuggling. This is the normal case of an empirical ML paper whose results are falsifiable against external benchmarks.
Axiom & Free-Parameter Ledger
axioms (2)
- domain assumption SE(3)-equivariance is required for physically consistent molecular representations
- domain assumption Electron density point clouds can be fused with discrete graphs to capture quantum behaviors missed by topology
Reference graph
Works this paper leans on
-
[1]
Electron density-enhanced molecular geometry learning
Hongxin Xiang, Jun Xia, Xin Jin, Wenjie Du, Li Zeng, and Xiangxiang Zeng. Electron density-enhanced molecular geometry learning. InProceedings of the Thirty-Fourth International Joint Conference on Artificial Intelligence, pages 7840–7848, 2025. doi: 10.24963/ijcai.2025/872
-
[2]
Msanchor: De novo molecular generation from mass spectrometry data with anchor-extended molecular scaffolds
Xiaohan Qin, Chao Wang, Zhengyang Zhou, Linjiang Chen, Wenjie Du, and Yang Wang. Msanchor: De novo molecular generation from mass spectrometry data with anchor-extended molecular scaffolds. In Proceedings of the AAAI Conference on Artificial Intelligence, volume 40, pages 953–961, 2026
2026
-
[3]
Jiajun Yu, Yizhen Zheng, Huan Yee Koh, Shirui Pan, Tianyue Wang, and Haishuai Wang. Collaborative expert llms guided multi-objective molecular optimization.arXiv preprint arXiv:2503.03503, 2025
-
[4]
Large language models for drug discovery and development.Patterns, 6(10), 2025
Yizhen Zheng, Huan Yee Koh, Jiaxin Ju, Madeleine Yang, Lauren T May, Geoffrey I Webb, Li Li, Shirui Pan, and George Church. Large language models for drug discovery and development.Patterns, 6(10), 2025
2025
-
[5]
Kun Li, Yida Xiong, Hongzhi Zhang, Xiantao Cai, Jia Wu, Bo Du, and Wenbin Hu. Graph-structured small molecule drug discovery through deep learning: Progress, challenges, and opportunities. In2025 IEEE International Conference on Web Services (ICWS), pages 1033–1042, 2025. doi: 10.1109/ICWS67624. 2025.00135
-
[6]
Can molecular evolution mechanism enhance molecular representation? InProceedings of the AAAI Conference on Artificial Intelligence, volume 40, pages 15108–15116, 2026
Kun Li, Longtao Hu, Jiameng Chen, Hongzhi Zhang, Yida Xiong, Xiantao Cai, Wenbin Hu, and Jia Wu. Can molecular evolution mechanism enhance molecular representation? InProceedings of the AAAI Conference on Artificial Intelligence, volume 40, pages 15108–15116, 2026
2026
-
[7]
Pcevo: Path-consistent molecular representation via virtual evolutionary
Kun Li, Longtao Hu, Yida Xiong, Jiajun Yu, Hongzhi Zhang, Jiameng Chen, Xiantao Cai, Jia Wu, and Wenbin Hu. Pcevo: Path-consistent molecular representation via virtual evolutionary. InProceedings of the Thirty-Fourth International Joint Conference on Artificial Intelligence, IJCAI-26. International Joint Conferences on Artificial Intelligence Organization...
2026
-
[8]
A centrality-based graph learning framework
Jiajun Yu, Zhihao Wu, Jielong Lu, Tianyue Wang, and Haishuai Wang. A centrality-based graph learning framework. InProceedings of the Thirty-Fourth International Joint Conference on Artificial Intelligence, pages 3588–3596, 2025
2025
-
[9]
Kenneth Atz, Francesca Grisoni, and Gisbert Schneider. Geometric deep learning on molecular representa- tions.Nature Machine Intelligence, 3(12):1023–1032, 2021. doi: 10.1038/s42256-021-00418-8
-
[10]
Zhen Li, Mingjian Jiang, Shuang Wang, and Shugang Zhang. Deep learning methods for molecular representation and property prediction.Drug Discovery Today, 27(12):103373, 2022. doi: 10.1016/j.drudis. 2022.103373
-
[11]
Kun Li, Zhennan Wu, Shoupeng Wang, Jia Wu, Shirui Pan, and Wenbin Hu. Drugpilot: Llm-based parameterized reasoning agent for drug discovery.arXiv preprint arXiv:2505.13940, 2025
-
[12]
Kun Li, Zhennan Wu, Yida Xiong, Hongzhi Zhang, Longtao Hu, Zhonglie Liu, Junqi Zeng, Wenjie Wu, Mukun Chen, Jiameng Chen, et al. Bsl: A unified and generalizable multitask learning platform for virtual drug discovery from design to synthesis.arXiv preprint arXiv:2508.01195, 2025
-
[13]
Smiles, a chemical language and information system
David Weininger. Smiles, a chemical language and information system. 1. introduction to methodology and encoding rules.Journal of Chemical Information and Computer Sciences, 28(1):31–36, 1988. doi: 10.1021/ci00057a005
-
[14]
Heller, Alan McNaught, Igor Pletnev, Stephen Stein, and Dmitrii Tchekhovskoi
Stephen R. Heller, Alan McNaught, Igor Pletnev, Stephen Stein, and Dmitrii Tchekhovskoi. Inchi, the iupac international chemical identifier.Journal of Cheminformatics, 7:23, 2015. doi: 10.1186/ s13321-015-0068-4
2015
-
[15]
Mario Krenn, Florian Häse, AkshatKumar Nigam, Pascal Friederich, and Alán Aspuru-Guzik. Self- referencing embedded strings (SELFIES): A 100% robust molecular string representation.Machine Learning: Science and Technology, 1(4):045024, 2020. doi: 10.1088/2632-2153/aba947
-
[16]
Kernel readout for graph neural networks
Jiajun Yu, Zhihao Wu, Jinyu Cai, Adele Lu Jia, and Jicong Fan. Kernel readout for graph neural networks. InIJCAI, pages 2505–2514, 2024
2024
-
[17]
Gotennet: Rethinking efficient 3d equivariant graph neural networks
Sarp Aykent and Tian Xia. Gotennet: Rethinking efficient 3d equivariant graph neural networks. InThe Thirteenth International Conference on Learning Representations, 2025. URL https://openreview. net/forum?id=5wxCQDtbMo. 10
2025
-
[18]
Yusong Wang, Tong Wang, Shaoning Li, Xinheng He, Mingyu Li, Zun Wang, Nanning Zheng, Bin Shao, and Tie-Yan Liu. Enhancing geometric representations for molecules with equivariant vector-scalar interactive message passing.Nature Communications, 15(1):313, 2024. doi: 10.1038/s41467-023-43720-2
-
[19]
Tensor field networks: Rotation- and translation-equivariant neural networks for 3D point clouds
Nathaniel Thomas, Tess Smidt, Steven Kearnes, Lusann Yang, Li Li, Kai Kohlhoff, and Patrick Riley. Tensor field networks: Rotation- and translation-equivariant neural networks for 3d point clouds.arXiv preprint arXiv:1802.08219, 2018
work page internal anchor Pith review Pith/arXiv arXiv 2018
-
[20]
Cormorant: Covariant molecular neural networks
Brandon Anderson, Truong-Son Hy, and Risi Kondor. Cormorant: Covariant molecular neural networks. InAdvances in Neural Information Processing Systems, volume 32, 2019
2019
-
[21]
E(n) equivariant graph neural networks
Victor Garcia Satorras, Emiel Hoogeboom, and Max Welling. E(n) equivariant graph neural networks. In Proceedings of the 38th International Conference on Machine Learning, volume 139 ofProceedings of Machine Learning Research, pages 9323–9332, 2021
2021
-
[22]
Wood, Abhishek Das, and Tess E
Yi-Lun Liao, Brandon M. Wood, Abhishek Das, and Tess E. Smidt. Equiformerv2: Improved equivariant transformer for scaling to higher-degree representations. InThe Twelfth International Conference on Learning Representations, 2024. URLhttps://openreview.net/forum?id=mCOBKZmrzD
2024
-
[23]
Inhomogeneous electron gas.Physical Review, 136(3B):B864–B871,
Pierre Hohenberg and Walter Kohn. Inhomogeneous electron gas.Physical Review, 136(3B):B864–B871,
-
[24]
doi: 10.1103/PhysRev.136.B864
-
[25]
W. Kohn and L. J. Sham. Self-consistent equations including exchange and correlation effects.Physical Review, 140(4A):A1133–A1138, 1965. doi: 10.1103/PhysRev.140.A1133
-
[26]
Parr and Weitao Yang.Density-Functional Theory of Atoms and Molecules, volume 16
Robert G. Parr and Weitao Yang.Density-Functional Theory of Atoms and Molecules, volume 16. Oxford University Press, New York, 1989
1989
-
[27]
Richard F. W. Bader. A quantum theory of molecular structure and its applications.Chemical Reviews, 91 (5):893–928, 1991. doi: 10.1021/cr00005a013
-
[28]
Johnson, Shahar Keinan, Paula Mori-Sánchez, Julia Contreras-García, Aron J
Erin R. Johnson, Shahar Keinan, Paula Mori-Sánchez, Julia Contreras-García, Aron J. Cohen, and Weitao Yang. Revealing noncovalent interactions.Journal of the American Chemical Society, 132(18):6498–6506,
-
[29]
doi: 10.1021/ja100936w
-
[30]
Zhuoran Qiao, Matthew Welborn, Animashree Anandkumar, Frederick R. Manby, and Thomas F. Miller III. Orbnet: Deep learning for quantum chemistry using symmetry-adapted atomic-orbital features.The Journal of Chemical Physics, 153(12):124111, 2020. doi: 10.1063/5.0021955
-
[31]
Peter Bjørn Jørgensen and Arghya Bhowmik. Deepdft: Neural message passing network for accurate charge density prediction.arXiv preprint arXiv:2011.03346, 2020
-
[32]
Equivariant graph neural networks for fast electron density estimation of molecules, liquids, and solids.npj Computational Materials, 8:183, 2022
Peter Bjørn Jørgensen and Arghya Bhowmik. Equivariant graph neural networks for fast electron density estimation of molecules, liquids, and solids.npj Computational Materials, 8:183, 2022. doi: 10.1038/ s41524-022-00863-y
2022
-
[33]
Sebastian Dick and Marivi Fernandez-Serra. Machine learning accurate exchange and correlation function- als of the electronic density.Nature Communications, 11:3509, 2020. doi: 10.1038/s41467-020-17265-7
-
[34]
He Li, Zun Wang, Nianlong Zou, Meng Ye, Runzhang Xu, Xiaoxun Gong, Wenhui Duan, and Yong Xu. Deep-learning density functional theory hamiltonian for efficient ab initio electronic-structure calculation. Nature Computational Science, 2(6):367–377, 2022. doi: 10.1038/s43588-022-00265-6
-
[35]
Dral, Matthias Rupp, and O
Raghunathan Ramakrishnan, Pavlo O. Dral, Matthias Rupp, and O. Anatole von Lilienfeld. Quantum chemistry structures and properties of 134 kilo molecules.Scientific Data, 1:140022, 2014. doi: 10.1038/ sdata.2014.22
2014
-
[36]
Qmugs, quantum mechanical properties of drug-like molecules.Scientific Data, 9(1):273, 2022
Clemens Isert, Kenneth Atz, José Jiménez-Luna, and Gisbert Schneider. Qmugs, quantum mechanical properties of drug-like molecules.Scientific Data, 9(1):273, 2022. doi: 10.1038/s41597-022-01390-7
-
[37]
Edbench: Large-scale electron density data for molecular modeling
Hongxin Xiang, Ke Li, Mingquan Liu, Zhixiang Cheng, Bin Yao, Wenjie Du, Jun Xia, Li Zeng, Xin Jin, and Xiangxiang Zeng. Edbench: Large-scale electron density data for molecular modeling. InAdvances in Neural Information Processing Systems, 2025. Datasets and Benchmarks Track
2025
-
[38]
Contrastive learning-based drug screening model for glun1/glun3a inhibitors.Acta Pharmacologica Sinica, pages 1–13, 2025
Kun Li, Yue Zeng, Yi-da Xiong, Hao-chen Wu, Sui Fang, Zhi-yan Qu, Yan Zhu, Bo Du, Zhao-bing Gao, and Wen-bin Hu. Contrastive learning-based drug screening model for glun1/glun3a inhibitors.Acta Pharmacologica Sinica, pages 1–13, 2025. 11
2025
-
[39]
Cohen, Paula Mori-Sánchez, and Weitao Yang
Aron J. Cohen, Paula Mori-Sánchez, and Weitao Yang. Challenges for density functional theory.Chemical Reviews, 112(1):289–320, 2012
2012
-
[40]
Perspective on density functional theory.The Journal of Chemical Physics, 136(15):150901, 2012
Kieron Burke. Perspective on density functional theory.The Journal of Chemical Physics, 136(15):150901, 2012
2012
-
[41]
Tanja Van Mourik, Michael Bühl, and Marie-Pierre Gaigeot. Density functional theory across chemistry, physics and biology.Philosophical Transactions of the Royal Society A: Mathematical, Physical and Engineering Sciences, 372(2011):20120488, 2014
2011
-
[42]
Nobel lecture: Electronic structure of matter—wave functions and density functionals
Walter Kohn. Nobel lecture: Electronic structure of matter—wave functions and density functionals. Reviews of Modern Physics, 71(5):1253–1266, 1999
1999
-
[43]
Schoenholz, Patrick F
Justin Gilmer, Samuel S. Schoenholz, Patrick F. Riley, Oriol Vinyals, and George E. Dahl. Neural message passing for quantum chemistry. InProceedings of the 34th International Conference on Machine Learning, volume 70 ofProceedings of Machine Learning Research, pages 1263–1272, 2017
2017
-
[44]
Convolutional networks on graphs for learning molecular fingerprints
David K Duvenaud, Dougal Maclaurin, Jorge Iparraguirre, Rafael Bombarell, Timothy Hirzel, Alan Aspuru-Guzik, and Ryan P Adams. Convolutional networks on graphs for learning molecular fingerprints. In C. Cortes, N. Lawrence, D. Lee, M. Sugiyama, and R. Garnett, editors,Advances in Neural Information Processing Systems, volume 28. Curran Associates, Inc., 2...
2015
-
[45]
Schütt, Pieter-Jan Kindermans, Huziel E
Kristof T. Schütt, Pieter-Jan Kindermans, Huziel E. Sauceda, Stefan Chmiela, Alexandre Tkatchenko, and Klaus-Robert Müller. Schnet: A continuous-filter convolutional neural network for modeling quantum interactions. InAdvances in Neural Information Processing Systems, volume 30, 2017
2017
-
[46]
Oliver T. Unke and Markus Meuwly. Physnet: A neural network for predicting energies, forces, dipole moments, and partial charges.Journal of Chemical Theory and Computation, 15(6):3678–3693, 2019. doi: 10.1021/acs.jctc.9b00181
-
[47]
Margraf, and Stephan Günnemann
Johannes Gasteiger, Shankari Giri, Johannes T. Margraf, and Stephan Günnemann. Fast and uncertainty- aware directional message passing for non-equilibrium molecules. InMachine Learning for Molecules Workshop, NeurIPS, 2020
2020
-
[48]
Spherical message passing for 3d molecular graphs
Yi Liu, Limei Wang, Meng Liu, Yuchao Lin, Xuan Zhang, Bora Oztekin, and Shuiwang Ji. Spherical message passing for 3d molecular graphs. InInternational Conference on Learning Representations (ICLR), 2022. URLhttps://openreview.net/forum?id=givsRXsOt9r
2022
-
[49]
Mailoa, Mordechai Kornbluth, Nicola Molinari, Tess E
Simon Batzner, Albert Musaelian, Lixin Sun, Mario Geiger, Jonathan P. Mailoa, Mordechai Kornbluth, Nicola Molinari, Tess E. Smidt, and Boris Kozinsky. E(3)-equivariant graph neural networks for data- efficient and accurate interatomic potentials.Nature Communications, 13(1):2453, 2022. doi: 10.1038/ s41467-022-29939-5
2022
-
[50]
Schütt, Oliver T
Kristof T. Schütt, Oliver T. Unke, and Michael Gastegger. Equivariant message passing for the prediction of tensorial properties and molecular spectra. InProceedings of the 38th International Conference on Machine Learning, volume 139 ofProceedings of Machine Learning Research, pages 9377–9388, 2021
2021
-
[51]
Qi, Hao Su, Kaichun Mo, and Leonidas J
Charles R. Qi, Hao Su, Kaichun Mo, and Leonidas J. Guibas. Pointnet: Deep learning on point sets for 3d classification and segmentation. InProceedings of the IEEE Conference on Computer Vision and Pattern Recognition, pages 77–85, 2017
2017
-
[52]
Qi, Li Yi, Hao Su, and Leonidas J
Charles R. Qi, Li Yi, Hao Su, and Leonidas J. Guibas. Pointnet++: Deep hierarchical feature learning on point sets in a metric space. InAdvances in Neural Information Processing Systems, volume 30, 2017
2017
-
[53]
Yue Wang, Yongbin Sun, Ziwei Liu, Sanjay E. Sarma, Michael M. Bronstein, and Justin M. Solomon. Dynamic graph cnn for learning on point clouds.ACM Transactions on Graphics, 38(5):146, 2019. doi: 10.1145/3326362
-
[54]
Hengshuang Zhao, Li Jiang, Jiaya Jia, Philip H. S. Torr, and Vladlen Koltun. Point transformer. In Proceedings of the IEEE/CVF International Conference on Computer Vision, pages 16259–16268, 2021
2021
-
[55]
Pointnext: Revisiting pointnet++ with improved training and scaling strategies
Guocheng Qian, Yuchen Li, Houwen Peng, Jinjie Mai, Hasan Hammoud, Mohamed Elhoseiny, and Bernard Ghanem. Pointnext: Revisiting pointnet++ with improved training and scaling strategies. InAdvances in Neural Information Processing Systems (NeurIPS), 2022
2022
-
[56]
Pointvector: A vector representation in point cloud analysis
Xin Deng, WenYu Zhang, Qing Ding, and XinMing Zhang. Pointvector: A vector representation in point cloud analysis. InProceedings of the IEEE/CVF Conference on Computer Vision and Pattern Recognition (CVPR), pages 9455–9465, 2023. 12
2023
-
[57]
Ismail Fawaz, H., Forestier, G., Weber, J., Idoumghar, L., and Muller, P.-A
Xiaoyang Wu, Li Jiang, Peng-Shuai Wang, Zhijian Liu, Xihui Liu, Yu Qiao, Wanli Ouyang, Tong He, and Hengshuang Zhao. Point transformer v3: Simpler, faster, stronger. InProceedings of the IEEE/CVF Conference on Computer Vision and Pattern Recognition (CVPR), pages 4840–4851, 2024. doi: 10.1109/CVPR52733.2024.00463
-
[58]
Psi4 1.4: Open-source software for high-throughput quantum chemistry.The Journal of Chemical Physics, 152(18), 2020
Daniel GA Smith, Lori A Burns, Andrew C Simmonett, Robert M Parrish, Matthew C Schieber, Raimondas Galvelis, Peter Kraus, Holger Kruse, Roberto Di Remigio, Asem Alenaizan, et al. Psi4 1.4: Open-source software for high-throughput quantum chemistry.The Journal of Chemical Physics, 152(18), 2020
2020
-
[59]
Gomez, Łukasz Kaiser, and Illia Polosukhin
Ashish Vaswani, Noam Shazeer, Niki Parmar, Jakob Uszkoreit, Llion Jones, Aidan N. Gomez, Łukasz Kaiser, and Illia Polosukhin. Attention Is All You Need. InAdvances in Neural Information Processing Systems, volume 30, pages 5998–6008, 2017
2017
-
[60]
Comenet: Towards complete and efficient message passing for 3d molecular graphs
Limei Wang, Yi Liu, Yuchao Lin, Haoran Liu, and Shuiwang Ji. Comenet: Towards complete and efficient message passing for 3d molecular graphs. InAdvances in Neural Information Processing Systems, volume 35, pages 650–664, 2022. URL https://proceedings.neurips.cc/paper_files/paper/ 2022/hash/0418973e545b932939302cb605d06f43-Abstract-Conference.html
-
[61]
Equiformer: Equivariant graph attention transformer for 3d atomistic graphs
Yi-Lun Liao and Tess Smidt. Equiformer: Equivariant graph attention transformer for 3d atomistic graphs. InInternational Conference on Learning Representations, 2023. URL https://openreview.net/ forum?id=KwmPfARgOTD. A Background and Theoretical Foundations A.1 Electron Density as a Molecular Quantum Field A.1.1 Definition of Electron Density Electron den...
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.