Electronic properties governing the phase stability and elastic anisotropy of C14 and C15 Cr-Hf-Nb Laves phases
Pith reviewed 2026-06-29 17:04 UTC · model grok-4.3
The pith
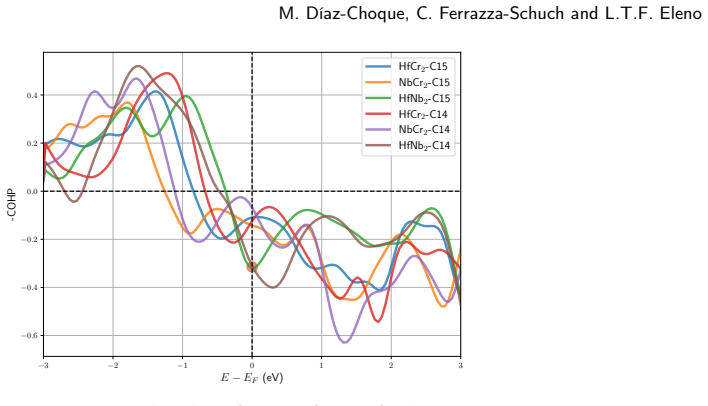
Strong anti-bonding near the Fermi level in XM2 M-M bonds destabilizes C14 and C15 Laves phases in the Cr-Hf-Nb system.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
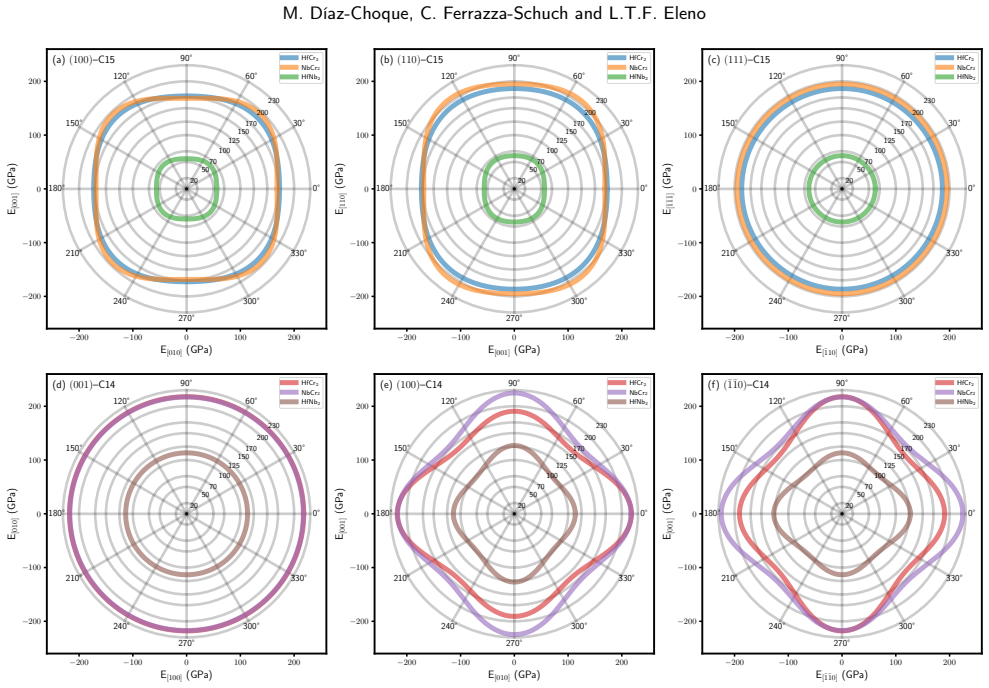
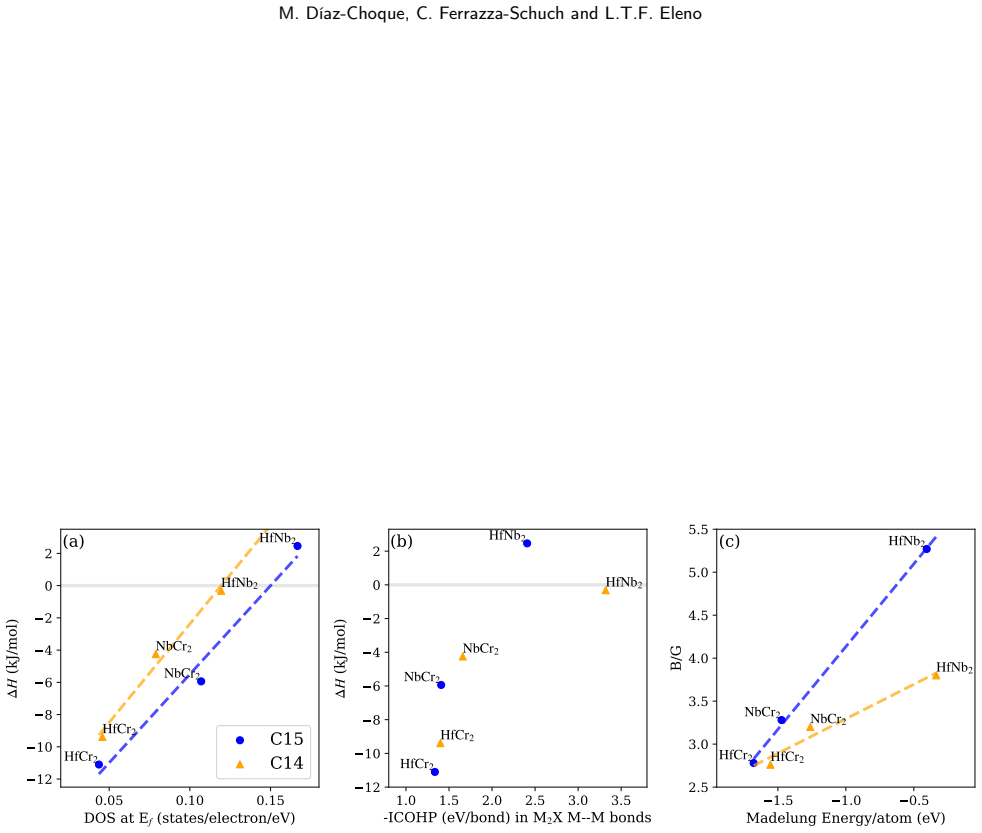
Both the C14 phases (HfNb2, HfCr2, NbCr2) and C15 phases (HfCr2, NbCr2) exhibit negative formation enthalpies and satisfy mechanical stability criteria. Elastic anisotropy decreases in the order HfCr2 > NbCr2 > HfNb2 for C14 but increases in the order NbCr2 > HfNb2 > HfCr2 for C15. COHP analysis reveals that strong anti-bonding behavior near the Fermi level within the XM2 M-M bonds serves as the primary mechanism destabilizing these Laves phases.
What carries the argument
Crystal orbital Hamilton population (COHP) analysis of the electronic density of states, highlighting anti-bonding states in the XM2 M-M bonds near the Fermi level.
Load-bearing premise
The DFT calculations with the chosen functional and parameters accurately represent the real electronic structure and bonding energies without significant systematic errors from approximations in the method or from the specific compositions studied.
What would settle it
A calculation or experiment showing positive COHP values (bonding) instead of negative (anti-bonding) near the Fermi level in XM2 M-M bonds would falsify the destabilization mechanism.
Figures
read the original abstract
This study utilizes Density Functional Theory (DFT) to investigate the thermodynamic stability, elastic anisotropy, and electronic properties of C14 and C15 Laves phases within the Cr--Hf--Nb system. Both formation enthalpies and comprehensive elastic property analyses confirm the energetic and mechanical stability of the C14 (HfNb$_2$, HfCr$_2$, NbCr$_2$) and C15 (HfCr$_2$, NbCr$_2$) phases. Furthermore, the evaluation of elastic anisotropy reveals a descending order of HfCr$_2$ > NbCr$_2$ > HfNb$_2$ for the C14 phase, contrasting with NbCr$_2$ > HfNb$_2$ > HfCr$_2$ for the C15 phase. Finally, electronic structure and COHP analyses indicate that strong anti-bonding behavior near the Fermi level within the XM$_2$ M--M bonds acts as a primary destabilization mechanism for both of these Laves phases.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript uses DFT to compute formation enthalpies, elastic constants, and anisotropy for C14 and C15 Laves phases (HfNb2, HfCr2, NbCr2) in the Cr-Hf-Nb system. Negative formation enthalpies and positive elastic moduli are taken to confirm thermodynamic and mechanical stability. Elastic anisotropy is ranked differently for the two structures. COHP analysis is used to identify strong anti-bonding character near EF in the XM2 M–M bonds as the primary electronic destabilization mechanism for both phases.
Significance. If the DFT results and COHP interpretation hold, the work supplies concrete stability and anisotropy data for ternary Laves phases relevant to refractory alloys. The linkage of elastic trends to specific bond types via COHP offers a mechanistic picture that could guide composition tuning, provided the electronic-structure conclusions survive functional-sensitivity tests.
major comments (2)
- [Abstract / electronic-structure analysis] Abstract and electronic-structure section: the claim that anti-bonding states in XM2 M–M bonds constitute the “primary destabilization mechanism” rests on a single GGA-level COHP analysis. Standard GGA functionals can shift d-band centers by 0.2–0.5 eV, which is sufficient to change whether anti-bonding states are occupied or to reorder their strength relative to X–M bonds; no hybrid-functional, GW, or experimental DOS comparison is reported to test this sensitivity.
- [Thermodynamic stability and elastic properties sections] Stability and elastic-property sections: while formation enthalpies and elastic constants are stated to confirm stability, the manuscript provides neither tabulated numerical values with error bars nor the specific exchange-correlation functional and convergence parameters used, preventing independent assessment of whether the reported ordering of phases is robust.
minor comments (2)
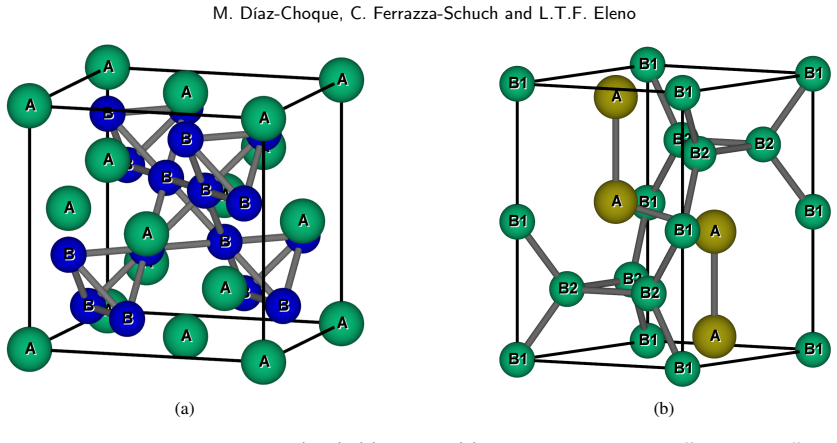
- [Introduction / methods] Notation for the XM2 M–M bonds should be defined explicitly on first use (e.g., which atoms occupy the X and M sites in each structure).
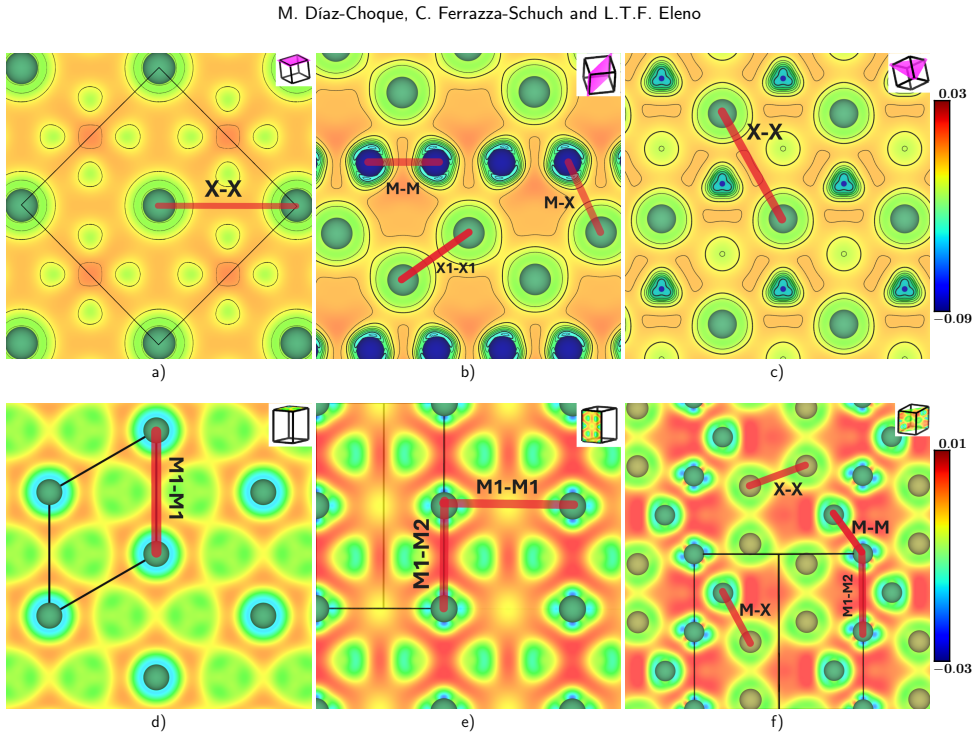
- [Figure captions] Figure captions for the COHP plots should state the integration range used to quantify “strong” anti-bonding and whether the plots are total or orbital-projected.
Simulated Author's Rebuttal
We thank the referee for the constructive comments on our manuscript. We address each major point below and have revised the manuscript to improve reproducibility and to qualify our electronic-structure claims appropriately.
read point-by-point responses
-
Referee: [Abstract / electronic-structure analysis] Abstract and electronic-structure section: the claim that anti-bonding states in XM2 M–M bonds constitute the “primary destabilization mechanism” rests on a single GGA-level COHP analysis. Standard GGA functionals can shift d-band centers by 0.2–0.5 eV, which is sufficient to change whether anti-bonding states are occupied or to reorder their strength relative to X–M bonds; no hybrid-functional, GW, or experimental DOS comparison is reported to test this sensitivity.
Authors: We acknowledge that the COHP analysis is performed at the GGA level and that functional sensitivity could in principle affect the precise positioning of anti-bonding states. The pronounced anti-bonding character near EF in the XM2 M–M bonds is nevertheless a robust qualitative feature of the calculated curves for all phases examined. In the revised manuscript we have changed the wording in the abstract and electronic-structure section from “primary destabilization mechanism” to “key destabilization mechanism” and added a short paragraph noting the known limitations of GGA for d-band positioning while emphasizing that the relative ordering of bond strengths remains consistent with prior GGA studies on related Laves phases. Additional hybrid-functional or GW calculations lie outside the computational scope of the present work. revision: partial
-
Referee: [Thermodynamic stability and elastic properties sections] Stability and elastic-property sections: while formation enthalpies and elastic constants are stated to confirm stability, the manuscript provides neither tabulated numerical values with error bars nor the specific exchange-correlation functional and convergence parameters used, preventing independent assessment of whether the reported ordering of phases is robust.
Authors: We agree that tabulated numerical values and full computational specifications are required for independent verification. The revised manuscript now includes explicit tables reporting the formation enthalpies and all independent elastic constants (with the values given to three decimal places), states that the PBE-GGA functional was employed, and lists the plane-wave cutoff, k-point meshes, and convergence thresholds used throughout the study. Because the results are deterministic DFT outputs, conventional statistical error bars are not applicable; the reported ordering of phases is therefore presented with the raw numerical data that allow direct assessment of robustness. revision: yes
Circularity Check
No circularity; direct DFT computations and COHP analysis are self-contained
full rationale
The paper's chain consists of standard DFT calculations for formation enthalpies, elastic constants, and COHP curves, from which stability, anisotropy ordering, and the anti-bonding interpretation are read out. No parameters are fitted to the target stability or anisotropy values, no self-definitional loops appear, and no load-bearing uniqueness theorems or ansatzes are imported via self-citation. The central claim follows directly from the computed electronic structure without reduction to its own inputs.
Axiom & Free-Parameter Ledger
axioms (1)
- domain assumption Density functional theory with a chosen exchange-correlation functional yields formation enthalpies and elastic constants accurate enough to determine relative phase stability and mechanical properties.
Reference graph
Works this paper leans on
-
[1]
R. L. Johnston, R. Hoffmann, Structure-bonding relationships in the Laves phases, Zeitschrift für anorganische und allgemeine Chemie 616 (10) (1992) 105–120. :Preprint submitted to Elsevier Page 9 of 11 M. Díaz-Choque, C. Ferrazza-Schuch and L.T.F. Eleno
1992
-
[2]
Paul-Boncour, S
V. Paul-Boncour, S. F. Matar, Ab initio approach of the hydrogen insertion effect on the magnetic properties of YFe2, Physical Review B—CondensedMatterandMaterialsPhysics70(18)(2004)184435
2004
-
[3]
N. Roy, K. MacIntosh, M. Eid, G. Canning, R. M. Rioux, The structural chemistry of intermetallic compounds enables active site design in heterogeneous catalysis, Chemical Science (2025)
2025
-
[4]
Ma, R.-K
L. Ma, R.-K. Pan, S.-C. Zhou, T.-P. Luo, D.-H. Wu, T.-W. Fan, B.-Y. Tang, Ab initio study of stacking faults and deformation mechanism inC15LavesphasesCr 2X(X=Nb,Zr,Hf),MaterialsChemistryand Physics 143 (2) (2014) 702–706
2014
-
[5]
Zhang, X
M. Zhang, X. Wang, M. Yang, M. Liang, J. Chen, Q. Zhou, P. Hou, H. Yu, F. Tian, First principles study of the structure and super- conductivity of La-Ge alloys under high pressure, Materials Today Communications 41 (2024) 110448
2024
-
[6]
Zhang, X
X. Zhang, X. Wang, B. Cao, C. Xing, F. Tian, First-principles study ofsuperconductivityinC15b–typeLaMAl 4 (M=Cl,Br,I,Te,Sc,Y, and Ce), Physics Letters A 564 (2025) 131122
2025
-
[7]
X. Ma, T. Wang, J. Wen, Z. Zhou, H. Zhu, First-principles study on structural,electronic,andsuperconductingpropertiesofLaves–phase alloyHfZn 2 underpressure,ChinesePhysicsB34(8)(2025)086108
2025
-
[8]
Koshinuma, H
T. Koshinuma, H. Ninomiya, I. Hase, H. Fujihisa, Y. Gotoh, K. Kawashima, S. Ishida, Y. Yoshida, H. Eisaki, T. Nishio, A. Iyo, High-pressure synthesis and superconductivity of the novel laves phase BaIr2, Intermetallics 148 (2022) 107643
2022
-
[9]
V. A. Yartys, M. V. Lototskyy, Laves type intermetallic compounds as hydrogen storage materials: A review, Journal of Alloys and Compounds 916 (2022) 165219
2022
-
[10]
J. B. Ponsoni, V. Aranda, T. da Silva Nascimento, R. B. Strozi, W. J. Botta, G. Zepon, Design of multicomponent alloys with C14 Lavesphasestructureforhydrogenstorageassistedbycomputational thermodynamic, Acta Materialia 240 (2022) 118317
2022
-
[11]
C. A. Paetsch, A. R. Natarajan, First-principles thermodynamics of hydrogen absorption in binary C15 Laves phases, Chemistry of Materials (2026)
2026
-
[12]
X.-Q. Chen, W. Wolf, R. Podloucky, P. Rogl, Ab initio study of ground-state properties of the Laves phase compounds TiCr2, ZrCr2, and HfCr2, Phys. Rev. B 71 (2005) 174101
2005
-
[13]
Schmetterer, A
C. Schmetterer, A. Khvan, A. Jacob, B. Hallstedt, T. Markus, A new theoreticalstudyoftheCr-Nbsystem,Journalofphaseequilibriaand diffusion 35 (2014) 434–444
2014
-
[14]
Q. Yao, J. Sun, D. Lin, S. Liu, B. Jiang, First-principles studies of defects, mechanical properties and electronic structure of Cr-based Laves phases, Intermetallics 15 (5) (2007) 694–699
2007
-
[15]
Z. Yang, M. Chisholm, B. Yang, X. Ma, Y. Wang, M. Zhuo, S. Pen- nycook, Role of crystal defects on brittleness of C15 Cr2Nb Laves phase, Acta Materialia 60 (6) (2012) 2637–2646
2012
-
[16]
L. A. Heaton, A. J. Samin, A first step towards understanding ther- momechanicalbehavioroftheNb-Crsystemthroughinteratomicpo- tential development and molecular dynamics simulations, Scientific Reports 14 (1) (2024) 14408
2024
-
[17]
S. Hong, C. Fu, Phase stability and elastic moduli of Cr2Nb by first- principles calculations, Intermetallics 7 (1) (1999) 5–9
1999
-
[18]
Bai, X.-R
H.-J. Bai, X.-R. Qin, First-principles study of the structural phase transition, elastic and thermodynamic properties of HfCr2, The Eu- ropean Physical Journal B 96 (5) (2023) 61
2023
-
[19]
Najrin, M
F. Najrin, M. A. Sarker, B. Neher, M. M. R. Bhuiyan, A comparative study of structural, elastic, electronic, thermophysical, and optical properties of cubic binary lave-phase intermetallic compounds of HfX2 (X= Cr, Mo, and W): An ab initio insight, Results in Materials 23 (2024) 100610
2024
-
[20]
L. Liu, P. Shen, X. Wu, R. Wang, W. Li, Q. Liu, First-principles cal- culations on the stacking fault energy, surface energy and dislocation propertiesofNbCr 2andHfCr 2,ComputationalMaterialsScience140 (2017) 334–343
2017
-
[21]
N. T. Hung, A. R. Nugraha, R. Saito, Quantum ESPRESSO course for solid-state physics, Jenny Stanford Publishing, 2022
2022
-
[22]
Giannozzi, S
P. Giannozzi, S. Baroni, N. Bonini, M. Calandra, R. Car, C. Cavaz- zoni, D. Ceresoli, G. L. Chiarotti, M. Cococcioni, I. Dabo, et al., Quantum espresso: a modular and open-source software project for quantumsimulationsofmaterials,Journalofphysics:Condensedmat- ter 21 (39) (2009) 395502
2009
-
[23]
Giannozzi, O
P. Giannozzi, O. Andreussi, T. Brumme, O. Bunau, M. B. Nardelli, M. Calandra, R. Car, C. Cavazzoni, D. Ceresoli, M. Cococcioni, et al., Advanced capabilities for materials modelling with quan- tum espresso, Journal of physics: Condensed matter 29 (46) (2017) 465901
2017
-
[24]
P.Hohenberg,W.Kohn,Inhomogeneouselectrongas,Physicalreview 136 (3B) (1964) B864
1964
-
[25]
W. Kohn, L. J. Sham, Self-consistent equations including exchange and correlation effects, Physical review 140 (4A) (1965) A1133
1965
-
[26]
J. P. Perdew, K. Burke, M. Ernzerhof, Generalized gradient approxi- mation made simple, Physical review letters 77 (18) (1996) 3865
1996
-
[27]
K. F. Garrity, J. W. Bennett, K. M. Rabe, D. Vanderbilt, Pseudopo- tentialsforhigh-throughputdftcalculations,ComputationalMaterials Science 81 (2014) 446–452
2014
-
[28]
URLhttps://link.aps.org/doi/10.1103/PhysRevLett.82.3296
N.Marzari,D.Vanderbilt,A.DeVita,M.C.Payne,Thermalcontrac- tionanddisorderingoftheal(110)surface,Phys.Rev.Lett.82(1999) 3296–3299.doi:10.1103/PhysRevLett.82.3296. URLhttps://link.aps.org/doi/10.1103/PhysRevLett.82.3296
-
[29]
H.J.Monkhorst,J.D.Pack,Specialpointsforbrillouin-zoneintegra- tions, Phys. Rev. B 13 (1976) 5188–5192
1976
-
[30]
A. D. Corso, The thermo_pw software,https://dalcorso.github.io/ thermo_pw
-
[31]
Z. Ran, C. Zou, Z. Wei, H. Wang, Velas: An open-source toolbox forvisualizationandanalysisofelasticanisotropy,ComputerPhysics Communications 283 (2023) 108540
2023
-
[32]
P. E. Blöchl, Projector augmented-wave method, Phys. Rev. B 50 (1994) 17953–17979
1994
-
[33]
Jollet, M
F. Jollet, M. Torrent, N. Holzwarth, Generation of projector augmented-wave atomic data: A 71 element validated table in the xmlformat,ComputerPhysicsCommunications185(4)(2014)1246– 1254
2014
-
[34]
P.E.Blöchl,O.Jepsen,O.K.Andersen,Improvedtetrahedronmethod forbrillouin-zoneintegrations,Phys.Rev.B49(1994)16223–16233. doi:10.1103/PhysRevB.49.16223. URLhttps://link.aps.org/doi/10.1103/PhysRevB.49.16223
-
[35]
R.Nelson,C.Ertural,J.George,V.L.Deringer,G.Hautier,R.Dron- skowski, LOBSTER: Local orbital projections, atomic charges, and chemical-bonding analysis from projector-augmented-wave- baseddensity-functionaltheory,JournalofComputationalChemistry 41 (21) (2020) 1931–1940
2020
-
[36]
Dronskowski, P
R. Dronskowski, P. E. Bloechl, Crystal orbital hamilton populations (COHP):energy-resolvedvisualizationofchemicalbondinginsolids based on density-functional calculations, The Journal of Physical Chemistry 97 (33) (1993) 8617–8624
1993
-
[37]
Momma, F
K. Momma, F. Izumi, VESTA: a three-dimensional visualization system for electronic and structural analysis, Journal of Applied Crystallography 41 (3) (2008) 653–658
2008
-
[38]
Birch, Equation of state and thermodynamic parameters of nacl to 300 kbar in the high-temperature domain, Journal of Geophysical Research: Solid Earth 91 (B5) (1986) 4949–4954
F. Birch, Equation of state and thermodynamic parameters of nacl to 300 kbar in the high-temperature domain, Journal of Geophysical Research: Solid Earth 91 (B5) (1986) 4949–4954
1986
-
[39]
F. Sun, J. Zhang, S. Mao, X. Han, First-principles studies of the structuralandelectronicpropertiesoftheC14LavesphaseXCr 2 (X= Ti,Zr,Nb,HfandTa),PhilosophicalMagazine93(19)(2013)2563– 2575
2013
-
[40]
Pavlů, J
J. Pavlů, J. Vřešt’ál, M. Šob, Thermodynamic modeling of Laves phases in the Cr–Hf and Cr–Ti systems: Reassessment using first- principles results, Calphad 34 (2) (2010) 215–221
2010
-
[41]
Villars, L
P. Villars, L. Calvert, Pearson’s handbook of crystallographic data, vol.1,TheMaterialsInformationSociety,MaterialsPark,OH(1991) 652
1991
-
[42]
Lu, W.-B
H.-J. Lu, W.-B. Wang, N. Zou, J.-Y. Shen, X.-G. Lu, Y.-L. He, ThermodynamicmodelingofCr–NbandZr–Crwithextensiontothe ternary Zr–Nb–Cr system, Calphad 50 (2015) 134–143. :Preprint submitted to Elsevier Page 10 of 11 M. Díaz-Choque, C. Ferrazza-Schuch and L.T.F. Eleno
2015
-
[43]
Hajra, C
R. Hajra, C. Panwisawas, J. Park, W. Choo, B. Han, J. Kim, High- temperaturephasestabilityandphasetransformationofNbCr 2 Laves phase:Experimentalandfirst-principlescalculationstudies,Materials and Design (2023)
2023
-
[44]
Trojko, et al., Structural investigations of the Nb1−𝑥Si𝑥T2 and Nb1−𝑥Al𝑥T2 (T= Cr, Mn, Fe, Co, Ni) systems, Journal of the Less Common Metals 119 (2) (1986) 297–305
R. Trojko, et al., Structural investigations of the Nb1−𝑥Si𝑥T2 and Nb1−𝑥Al𝑥T2 (T= Cr, Mn, Fe, Co, Ni) systems, Journal of the Less Common Metals 119 (2) (1986) 297–305
1986
-
[45]
F. Chu, D. Thoma, Y. He, T. Mitchell, S. Chen, J. Petepezki, Theo- retical and experimental studies on the C15 intermetallic compound NbCr2, MRS Online Proceedings Library (OPL) 364 (1994) 1089
1994
-
[46]
Martin, F
J. Martin, F. Müller, O. Kubaschewski, Thermodynamic properties of TaCr2 and NbCr2, Transactions of the Faraday Society 66 (1970) 1065–1072
1970
-
[47]
F. Sun, J. Zhang, S. Mao, X. Han, Structural, electronic and elastic properties of the C14 NbCr2 Laves phase under hydrostatic pressure, Solid state communications 174 (2013) 46–49
2013
-
[48]
Murad, Z
M. Murad, Z. Ali, Comprehensive dft exploration of structural, elec- tronic,magneticandelasticpropertiesofHfM 2 (M=Cr,Mn,andFe) intermetallic compounds, Physica B: Condensed Matter 675 (2024) 415641
2024
-
[49]
C. Li, B. Wang, Y. Li, R. Wang, First-principles study of the ideal cleavage fracture of Cr2Nb microalloyed by X (Al, Ni, Co, Ti), Intermetallics 17 (6) (2009) 394–399
2009
-
[50]
Thoma, F
D. Thoma, F. Chu, P. Peralta, P. Kotula, K. Chen, T. Mitchell, Elastic andmechanicalpropertiesofNb(Cr,V) 2C15Lavesphases,Materials Science and Engineering: A 239 (1997) 251–259
1997
-
[51]
H.Huang,G.Li,X.Xiao,S.Lu,P.Peng,Micromechanisminfracture toughness of NbCr2 Laves phase improved by nickel alloying: First- principles calculation, Journal of Alloys and Compounds 857 (2021) 158040
2021
-
[52]
F. Chu, Y. He, D. Thoma, T. Mitchell, Elastic constants of the C15 Laves phase compound NbCr2, Scripta metallurgica et materialia 33 (8) (1995)
1995
-
[53]
Hichour, D
M. Hichour, D. Rached, R. Khenata, M. Rabah, M. Merabet, A. H. Reshak, S. B. Omran, R. Ahmed, Theoretical investigations of nitisn and covsn compounds, Journal of Physics and Chemistry of Solids 73 (8) (2012) 975–981
2012
-
[54]
Born, On the stability of crystal lattices
M. Born, On the stability of crystal lattices. I, in: Mathematical Proceedings of the Cambridge Philosophical Society, Vol. 36, 1940, pp. 160–172
1940
-
[55]
A.Reuss,Z.Angnew,Acalculationofthebulkmodulusofpolycrys- talline materials, Math Meth 9 (1929) 55
1929
-
[56]
Voigt, Lehrbuch der kristallphysik:(mit ausschluss der kristallop- tik), Vol
W. Voigt, Lehrbuch der kristallphysik:(mit ausschluss der kristallop- tik), Vol. 34, BG Teubner, 1910
1910
-
[57]
Hill, The elastic behaviour of a crystalline aggregate, Proceedings of the Physical Society
R. Hill, The elastic behaviour of a crystalline aggregate, Proceedings of the Physical Society. Section A 65 (5) (1952) 349
1952
-
[58]
J.Qi,Y.Zhou,W.Wang,L.Qian,Z.Lv,W.Fu,Electronic,magnetic and mechanical properties of (Fe, Ni)2Nb from density functional theory, Journal of Magnetism and Magnetic Materials 452 (2018) 219–229
2018
-
[59]
Y.Tian,B.Xu,Z.Zhao,Microscopictheoryofhardnessanddesignof novel superhard crystals, International Journal of Refractory Metals and Hard Materials 33 (2012) 93–106
2012
-
[60]
O. L. Anderson, A simplified method for calculating the debye tem- perature from elastic constants, Journal of Physics and Chemistry of Solids 24 (7) (1963) 909–917
1963
-
[61]
S.Pugh,D.Edinburgh,Magazine,joscience,xcii,Relationsbetween the elastic moduli and the plastic properties of polycrystalline pure metals 45 (1954) 823–843
1954
-
[62]
E.Tindibale,W.M.Mulwa,B.I.Adetunji,Elastic,anisotropic,lattice dynamicsandelectronicpropertiesofXNiMandXNi 2M(X=Ti,Zr, Hf; M= Sn, Ge, Si): Dft comparison study, Physica B: Condensed Matter 665 (2023) 415029
2023
-
[63]
Y. Fang, Y. Wang, H. Imtiaz, B. Liu, H. Gao, Energy-ratio-based measure of elastic anisotropy, Physical review letters 122 (4) (2019) 045502
2019
-
[64]
C. M. Zener, S. Siegel, Elasticity and anelasticity of metals., The Journal of Physical Chemistry 53 (9) (1949) 1468–1468
1949
-
[65]
S.-C. Wu, G. H. Fecher, S. Shahab Naghavi, C. Felser, Elastic prop- erties and stability of heusler compounds: Cubic Co2YZ compounds with L21structure, Journal of Applied Physics 125 (8) (2019)
2019
-
[66]
S.I.Ranganathan,M.Ostoja-Starzewski,Universalelasticanisotropy index, Phys. Rev. Lett. 101 (2008) 055504
2008
-
[67]
D. H. Chung, W. R. Buessem, The elastic anisotropy of crystals, Journal of Applied Physics 38 (5) (1967) 2010–2012
1967
-
[68]
Ravindran, L
P. Ravindran, L. Fast, P. A. Korzhavyi, B. Johansson, J. Wills, O. Eriksson, Density functional theory for calculation of elastic properties of orthorhombic crystals: Application to TiSi2, Journal of Applied Physics 84 (9) (1998) 4891–4904
1998
-
[69]
Maxisch, G
T. Maxisch, G. Ceder, Elastic properties of olivine Li𝑥FePO4 from first principles, Phys. Rev. B 73 (2006) 174112
2006
-
[70]
Z.Ran,C.Zou,Z.Wei,H.Wang,R.Zhang,N.Fang,Phasetransitions and elastic anisotropies of SiC polymorphs under high pressure, Ceramics International 47 (5) (2021) 6187–6200
2021
-
[71]
B. Liu, G. Li, X. Xiao, S. Lu, P. Peng, Alloying effect on the mechanicalpropertiesofLavesphaseNbCr 2:Afirst-principlesstudy, Computational Materials Science 218 (2023) 111949
2023
-
[72]
V. L. Deringer, A. L. Tchougréeff, R. Dronskowski, Crystal orbital hamilton population (COHP) analysis as projected from plane-wave basis sets, The Journal of Physical Chemistry A 115 (21) (2011) 5461–5466
2011
-
[73]
S.Steinberg,R.Dronskowski,Thecrystalorbitalhamiltonpopulation (cohp) method as a tool to visualize and analyze chemical bonding in intermetallic compounds, Crystals 8 (5) (2018).doi:10.3390/ cryst8050225
2018
-
[74]
Ertural, S
C. Ertural, S. Steinberg, R. Dronskowski, Development of a robust tool to extract mulliken and löwdin charges from plane waves and its applicationtosolid-statematerials,RSCAdv.9(2019)29821–29830
2019
-
[75]
Bunge, J
C. Bunge, J. Barrientos, A. Bunge, Roothaan-hartree-fock ground- state atomic wave functions: Slater-type orbital expansions and ex- pectation values for Z = 2-54, Atomic Data and Nuclear Data Tables 53 (1) (1993) 113–162
1993
-
[76]
A. Landa, P. Söderlind, A. V. Ruban, O. E. Peil, L. Vitos, Stability inbcctransitionmetals:Madelungandband-energyeffectsduetoal- loying,Phys.Rev.Lett.103(2009)235501.doi:10.1103/PhysRevLett. 103.235501. :Preprint submitted to Elsevier Page 11 of 11 M. Díaz-Choque, C. Ferrazza-Schuch and L.T.F. Eleno □3 □2 □1 0 1 2 3 E □ EF (eV) □0.6 □0.4 □0.2 0.0 0.2 0.4 ...
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.