Rapid estimation of synthesizability windows of inorganic materials from first principles
Pith reviewed 2026-06-29 16:50 UTC · model grok-4.3
The pith
Machine-learned interatomic potentials enable rapid generation of predominance diagrams that predict synthesizability windows for inorganic materials.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
By combining density functional theory with machine-learned interatomic potentials, the authors generate phase predominance diagrams as a function of temperature and partial pressures for binary compounds and 48 ternary metal phosphosulfide systems. These diagrams show good agreement with experimental synthesis literature and identify synthesis windows where compounds that are metastable on a zero-temperature stability hull become thermodynamically stable.
What carries the argument
Phase predominance diagrams computed from free energies obtained via a hybrid DFT and machine-learned interatomic potential method, plotted against temperature and partial pressures of gaseous reactants.
If this is right
- Experimentalists can directly translate the diagrams into lab synthesis parameters for temperature and gas pressures.
- The method reduces computational cost drastically relative to a full DFT approach.
- Compounds that appear metastable on zero-temperature stability hulls can be thermodynamically stable under well-defined synthesis windows.
- The approach applies to binary compounds and scales to ternary systems such as metal phosphosulfides.
- The diagrams provide a practical bridge between computational stability predictions and experimental realization.
Where Pith is reading between the lines
- Routine use of such diagrams could shift materials screening workflows to prioritize candidates with accessible synthesis windows rather than zero-temperature stability alone.
- The method might be extended to quaternary or higher-order systems and to other synthesis variables such as solvent effects.
- Discrepancies between diagram predictions and experiments could highlight cases where kinetic barriers, rather than thermodynamics, control outcomes.
Load-bearing premise
The machine-learned interatomic potentials accurately capture the thermodynamics at finite temperature and pressure, and that reaching thermodynamic equilibrium is sufficient for experimental synthesizability.
What would settle it
An experimental attempt to synthesize one of the studied compounds under conditions the diagram marks as stable that fails to produce the phase, or success under conditions marked as unstable.
Figures

read the original abstract
Fast prediction of the synthesizability conditions of materials remains challenging, even assuming synthesis under thermodynamic equilibrium. We combine density functional theory (DFT) with machine-learned interatomic potentials to enable high-throughput generation of phase predominance diagrams as a function of temperature and partial pressures of the gaseous reactants. These diagrams can immediately be used by experimentalists to translate computational predictions into real synthesis parameters in the lab. Predominance diagrams are generated for a diverse set of binary compounds and for 48 more complex ternary metal phosphosulfide systems, but the method is in principle scalable to any inorganic material class. The calculated predominance diagrams generally show good agreement with the experimental synthesis literature, with a drastic reduction in computational cost compared to a full DFT approach. We find several examples of compounds that appear as metastable in a zero-temperature stability hull picture, but that become thermodynamically stable under well-defined synthesis windows.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript presents a hybrid DFT + machine-learned interatomic potential workflow to construct temperature- and partial-pressure-dependent phase predominance diagrams for inorganic compounds. The approach is demonstrated on a set of binary materials and on 48 ternary metal phosphosulfide systems; the resulting diagrams are reported to agree generally with experimental synthesis conditions, to identify cases in which phases that are metastable on the 0 K convex hull become thermodynamically stable inside finite-T/P windows, and to achieve a large reduction in computational cost relative to a pure DFT treatment.
Significance. If the validation of the ML potentials and the mapping from equilibrium diagrams to experimental outcomes hold, the work would provide a practical, high-throughput route for translating first-principles stability predictions into laboratory synthesis parameters. The ability to recover synthesis windows for selected metastable phases and the demonstrated application to 48 ternary systems are notable strengths that could accelerate materials discovery pipelines.
major comments (2)
- [Results] Results section: the statement that the diagrams 'generally show good agreement with the experimental synthesis literature' is not accompanied by quantitative metrics (e.g., fraction of systems for which the predicted window overlaps the reported experimental conditions, mean deviation in temperature or pressure, or size and composition of the validation set). Without these numbers it is difficult to judge whether the central claim of predictive utility is supported.
- [Methods] Methods section: the accuracy of the machine-learned potentials for the finite-temperature free energies and chemical potentials that enter the predominance diagrams is not benchmarked against DFT or experiment for the specific ternary systems studied. Because this accuracy is load-bearing for the reported stability windows, explicit error bars or cross-validation statistics are required.
minor comments (2)
- [Abstract] The abstract would be strengthened by inclusion of at least one quantitative performance figure (e.g., 'agreement in 42 of 48 systems within 50 K').
- Figure captions for the predominance diagrams should explicitly state the reference states and the range of partial pressures examined.
Simulated Author's Rebuttal
We thank the referee for the positive evaluation and the constructive comments on validation. We address each major comment below and will revise the manuscript accordingly.
read point-by-point responses
-
Referee: [Results] Results section: the statement that the diagrams 'generally show good agreement with the experimental synthesis literature' is not accompanied by quantitative metrics (e.g., fraction of systems for which the predicted window overlaps the reported experimental conditions, mean deviation in temperature or pressure, or size and composition of the validation set). Without these numbers it is difficult to judge whether the central claim of predictive utility is supported.
Authors: We agree that quantitative metrics are needed to support the claim of agreement. In the revised manuscript we will add a table in the Results section that, for all 48 ternary systems, indicates whether the computed predominance window overlaps the experimental synthesis conditions reported in the literature, together with the overall fraction of systems showing overlap and mean deviations in temperature and pressure where data permit. revision: yes
-
Referee: [Methods] Methods section: the accuracy of the machine-learned potentials for the finite-temperature free energies and chemical potentials that enter the predominance diagrams is not benchmarked against DFT or experiment for the specific ternary systems studied. Because this accuracy is load-bearing for the reported stability windows, explicit error bars or cross-validation statistics are required.
Authors: We acknowledge that explicit benchmarking of the ML potentials on the ternary systems is required. In the revised Methods section we will report cross-validation statistics (MAE and RMSE) for finite-temperature free energies and chemical potentials on a held-out subset of the ternary compounds, obtained by direct comparison to additional DFT calculations, and will include error bars on the reported stability windows. revision: yes
Circularity Check
No significant circularity detected
full rationale
The derivation relies on standard DFT combined with external machine-learned interatomic potentials to compute predominance diagrams as a function of T and partial pressures, followed by direct comparison to experimental synthesis literature for 48 ternary systems. No load-bearing step reduces by construction to a fitted parameter, self-definition, or self-citation chain; the validation is performed against independent external data rather than internal targets. The method is presented as scalable and cost-reducing without invoking uniqueness theorems or ansatzes from prior author work.
Axiom & Free-Parameter Ledger
Reference graph
Works this paper leans on
-
[1]
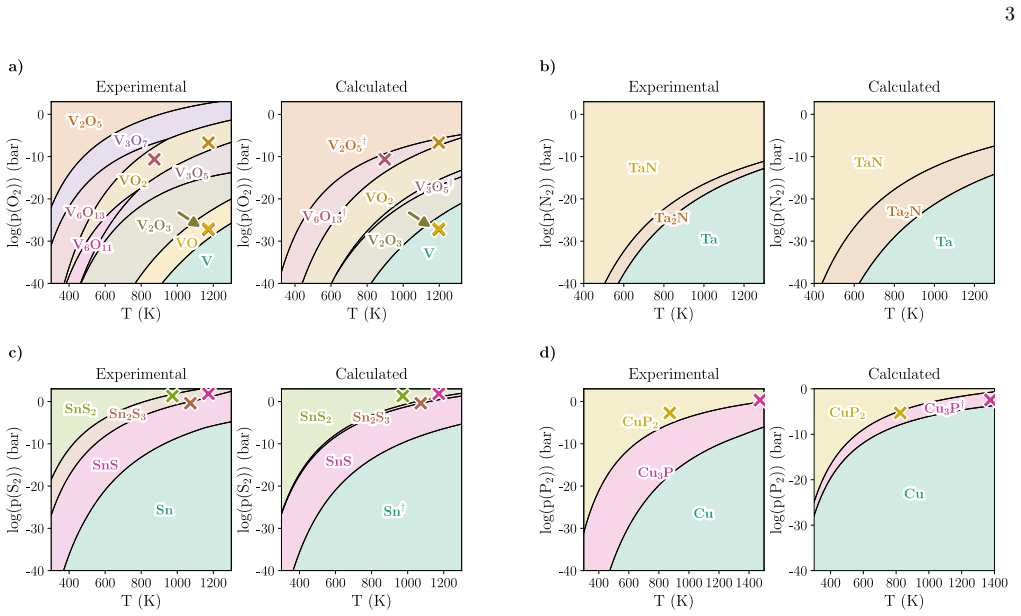
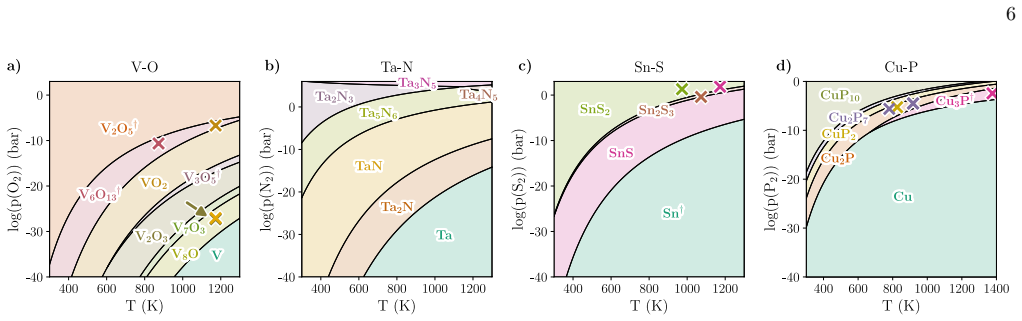
Errors and uncertainty Phase predominance diagrams showing compound sta- bility regions as a function of temperature and partial pressure of the reactant gas are constructed for four chosen binary systems in the oxide, nitride, sulfide and phosphide families (Fig. 2). The specific binary sys- tems (V-O, Ta-N, Sn-S, and Cu-P) were chosen as they contain a ...
-
[2]
Only compounds in- cluded in both our chosen experimental thermochemical database (HSC Chemistry [26]) and our chosen compu- tational database of DFT formation enthalpies (Ref
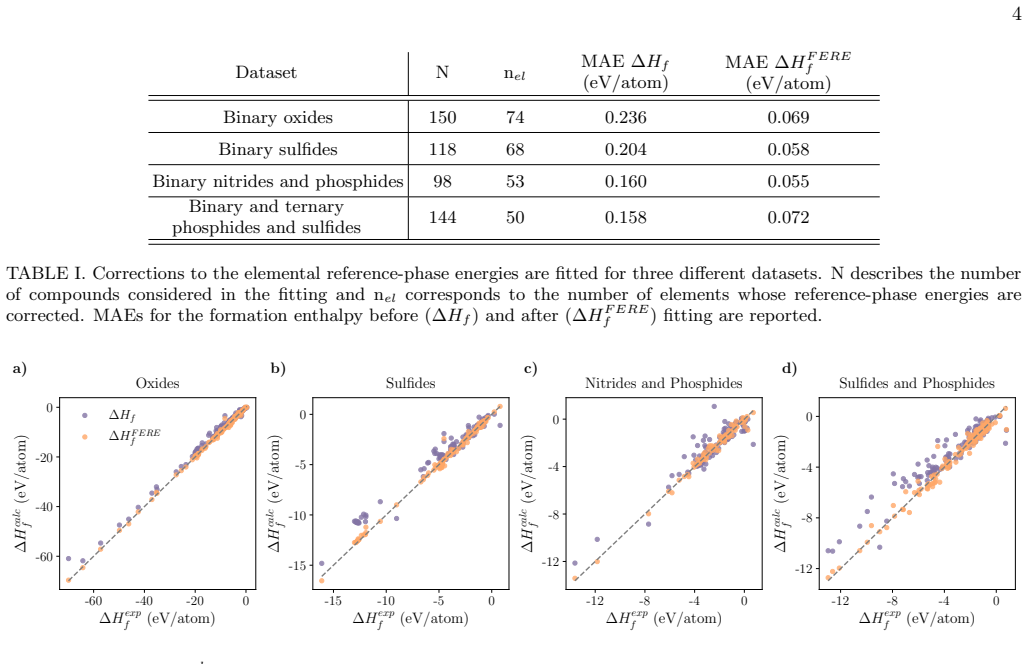
Comparison with experiment To assess the impact of these uncertainties on the pre- dicted phase predominance diagrams, phase boundaries derived using the workflow presented in this work are compared to phase diagrams constructed solely from ex- perimental thermochemical data. Only compounds in- cluded in both our chosen experimental thermochemical databas...
-
[3]
We therefore construct a new set of pre- dominance diagrams (Fig
Predictive power After assessing our method’s limitations and its perfor- mance against experimental data, we now turn to using it as a predictive tool to identify synthesis conditions for new materials. We therefore construct a new set of pre- dominance diagrams (Fig. 5) by considering all materials that are present either on Materials Project or the ICS...
1950
-
[4]
The chemical space of interest is ternary metal phosphosulfides including (almost) all metals from the s, p and d-block
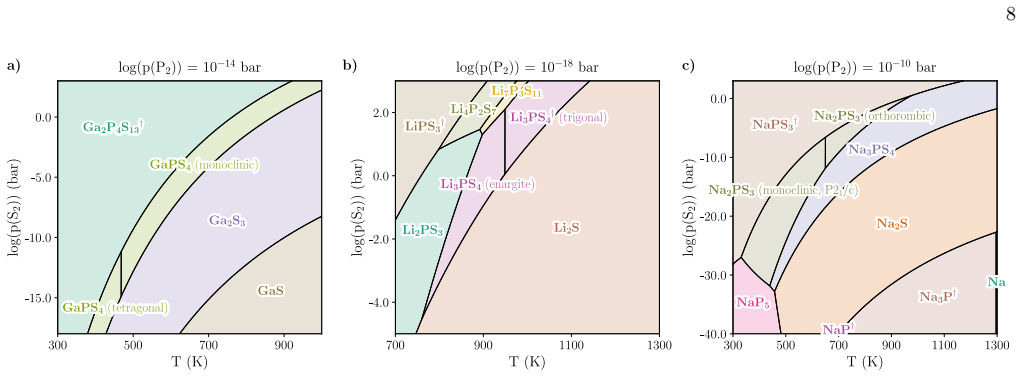
High-throughput predominance diagrams As a second case study, we show that our proposed method can readily be scaled to more than 1000 ma- terials, even in the more complex case of multinary systems where two elements are supplied from the gas phase. The chemical space of interest is ternary metal phosphosulfides including (almost) all metals from the s, ...
-
[5]
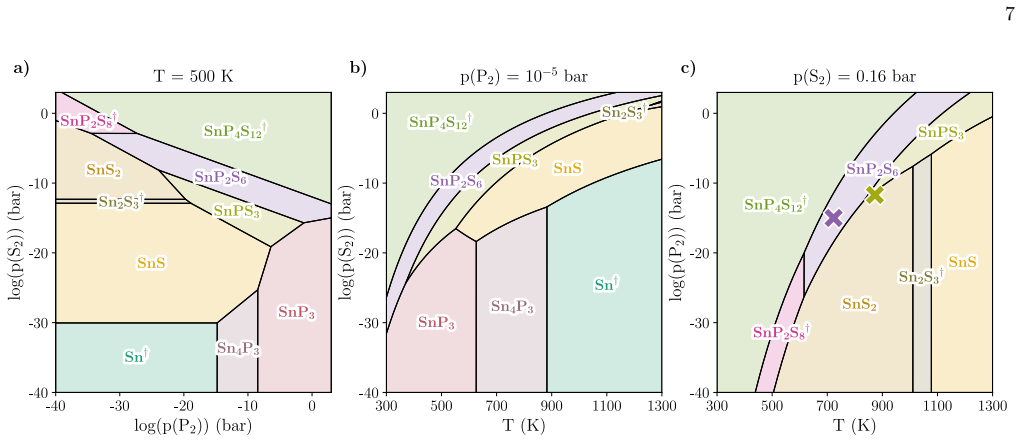
The Sn-P-S ternary system as a case study A variety of representations of the phase predominance diagrams are possible for ternary phosphosulfides, owing to the multivariate dependence of the Gibbs free energy on temperature and partial pressures of the two assumed reactant gases (P 2 and S 2). Fig. 6 a) shows phase pre- dominance in the Sn-P-S system as ...
-
[6]
They can be attributed to differences in the temperature dependence of thermochemical properties in the polymorphs, leading to reordering of their Gibbs free energies
Predicting phase transitions Structural phase transitions between different poly- morphs with the same composition can be predicted by our method. They can be attributed to differences in the temperature dependence of thermochemical properties in the polymorphs, leading to reordering of their Gibbs free energies. For our ternary phosphosulfide dataset, ph...
-
[7]
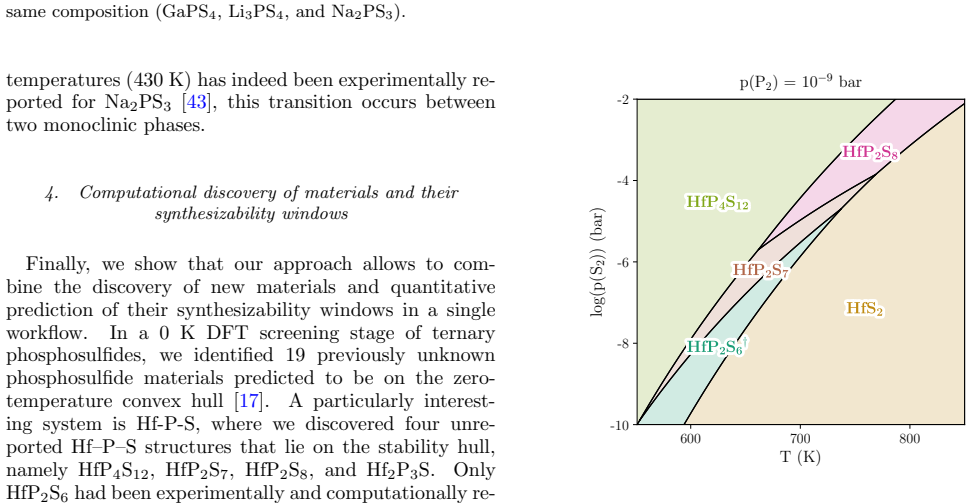
In a 0 K DFT screening stage of ternary phosphosulfides, we identified 19 previously unknown phosphosulfide materials predicted to be on the zero- temperature convex hull [17]
Computational discovery of materials and their synthesizability windows Finally, we show that our approach allows to com- bine the discovery of new materials and quantitative prediction of their synthesizability windows in a single workflow. In a 0 K DFT screening stage of ternary phosphosulfides, we identified 19 previously unknown phosphosulfide materia...
-
[8]
A. Jain, S. P. Ong, G. Hautier, W. Chen, W. D. Richards, S. Dacek, S. Cholia, D. Gunter, D. Skinner, G. Ceder, and K. A. Persson, Commentary: The Materials Project: A materials genome approach to accelerating materials innovation, APL Materials1, 011002 (2013)
2013
-
[9]
Kirklin, J
S. Kirklin, J. E. Saal, B. Meredig, A. Thompson, J. W. Doak, M. Aykol, S. R¨ uhl, and C. Wolverton, The Open Quantum Materials Database (OQMD): Assessing the accuracy of DFT formation energies, npj Computational Materials1, 15010 (2015)
2015
-
[10]
Schmidt, N
J. Schmidt, N. Hoffmann, H.-C. Wang, P. Borlido, P. J. M. A. Carri¸ co, T. F. T. Cerqueira, S. Botti, and M. A. L. Marques, Machine-Learning-Assisted Determination of the Global Zero-Temperature Phase Diagram of Mate- rials, Advanced Materials (Deerfield Beach, Fla.)35, e2210788 (2023)
2023
-
[11]
Kayastha, G
P. Kayastha, G. Longo, and L. D. Whalley, A First- Principles Thermodynamic Model for the Ba–Zr–S Sys- tem in Equilibrium with Sulfur Vapor, ACS Applied En- ergy Materials7, 11326 (2024)
2024
-
[12]
Zunger, S.-H
A. Zunger, S.-H. Wei, L. G. Ferreira, and J. E. Bernard, Special quasirandom structures, Physical Review Letters 65, 353 (1990)
1990
-
[13]
J. J. Cordell, J. Pan, A. C. Tamboli, G. J. Tucker, and S. Lany, Probing configurational disorder in ZnGeN 2 us- ing cluster-based Monte Carlo, Physical Review Materi- als5, 024604 (2021)
2021
-
[14]
C. J. Bartel, S. L. Millican, A. M. Deml, J. R. Rumptz, W. Tumas, A. W. Weimer, S. Lany, V. Stevanovi´ c, C. B. Musgrave, and A. M. Holder, Physical descrip- tor for the Gibbs energy of inorganic crystalline solids and temperature-dependent materials chemistry, Nature Communications9, 4168 (2018)
2018
-
[15]
Sun and N
W. Sun and N. David, A critical reflection on attempts to machine-learn materials synthesis insights from text- mined literature recipes, Faraday Discussions256, 614 (2025)
2025
-
[16]
Thermodynamic assessment of machine learning models for solid-state synthesis prediction
J. Schlesinger, S. Hjaltason, N. J. Szymanski, and C. J. Bartel, Thermodynamic assessment of machine learning models for solid-state synthesis prediction, https://arxiv.org/abs/2602.04075v1 (2026)
work page internal anchor Pith review Pith/arXiv arXiv 2026
-
[17]
Tolborg and A
K. Tolborg and A. Walsh, Low-Cost Vibrational Free En- ergies in Solid Solutions with Machine Learning Force Fields, The Journal of Physical Chemistry Letters14, 11618 (2023)
2023
-
[18]
S. Zhu, D. Sarıt¨ urk, and R. Arr´ oyave, Machine learn- ing potentials for alloys: A detailed workflow to predict phase diagrams and benchmark accuracy, npj Computa- tional Materials11, 340 (2025)
2025
-
[19]
Unglert, M
N. Unglert, M. Ketter, and G. K. H. Madsen, Active learning potentials for first-principles phase diagrams us- ing replica-exchange nested sampling, npj Computational Materials12, 107 (2026)
2026
-
[20]
H. Yang, C. Hu, Y. Zhou, X. Liu, Y. Shi, J. Li, G. Li, Z. Chen, S. Chen, C. Zeni, M. Horton, R. Pinsler, A. Fowler, D. Z¨ ugner, T. Xie, J. Smith, L. Sun, Q. Wang, L. Kong, C. Liu, H. Hao, and Z. Lu, MatterSim: A Deep Learning Atomistic Model Across Elements, Tempera- tures and Pressures (2024), arXiv:2405.04967 [cond-mat]
work page internal anchor Pith review Pith/arXiv arXiv 2024
-
[21]
A. Loew, D. Sun, H.-C. Wang, S. Botti, and M. A. L. Marques, Universal machine learning interatomic poten- tials are ready for phonons, npj Computational Materials 12 11, 178 (2025)
2025
-
[22]
Comparotto, L
C. Comparotto, L. Whalley, K. Sopiha, R. J. W. Frost, T. Kubart, and J. J. S. Scragg, Thermodynamic in- sights into the Ba–S system for the formation of BaZrS3 perovskites and other Ba sulfides, Journal of Materials Chemistry A13, 9983 (2025)
2025
-
[23]
Stevanovi´ c, S
V. Stevanovi´ c, S. Lany, X. Zhang, and A. Zunger, Cor- recting density functional theory for accurate predictions of compound enthalpies of formation: Fitted elemental- phase reference energies, Physical Review B85, 115104 (2012)
2012
- [24]
-
[25]
Pallikara, P
I. Pallikara, P. Kayastha, J. M. Skelton, and L. D. Whal- ley, The physical significance of imaginary phonon modes in crystals, Electronic Structure4, 033002 (2022)
2022
-
[26]
L. A. Burton, D. Colombara, R. D. Abellon, F. C. Grozema, L. M. Peter, T. J. Savenije, G. Dennler, and A. Walsh, Synthesis, Characterization, and Electronic Structure of Single-Crystal SnS, Sn2S3, and SnS2, Chem- istry of Materials25, 4908 (2013)
2013
-
[27]
Mootz and H
D. Mootz and H. Puhl, Die Kristallstruktur von Sn2S3, Acta Crystallographica23, 471 (1967)
1967
-
[28]
J. P. Odile, S. Soled, C. A. Castro, and A. Wold, Crystal growth and characterization of the transition- metal phosphides copper diphosphide, nickel diphos- phide, and rhodium triphosphide, Inorganic Chemistry 17, 283 (1978)
1978
-
[29]
Nowotny and E
H. Nowotny and E. Henglein, Ein Beitrag zur Kenntnis tern¨ arer Phosphorlegierungen, Monatshefte f¨ ur Chemie und verwandte Teile anderer Wissenschaften79, 385 (1948)
1948
-
[30]
Westman, I
S. Westman, I. Lindqvist, B. Sparrman, G. B. Nielsen, H. Nord, and A. Jart, Note on a Phase Transition in VO2., Acta Chemica Scandinavica15, 217 (1961)
1961
-
[31]
Aebi, Phasenuntersuchungen im System Vanadin- Sauerstoff und die Krystallstruktur von V12O26, Hel- vetica Chimica Acta31, 8 (1948)
F. Aebi, Phasenuntersuchungen im System Vanadin- Sauerstoff und die Krystallstruktur von V12O26, Hel- vetica Chimica Acta31, 8 (1948)
1948
-
[32]
J. A. Pople, Quantum Chemical Models (Nobel Lecture), Angewandte Chemie38, 1894 (1999)
1999
-
[33]
Roine, HSC Chemistry, Metso
A. Roine, HSC Chemistry, Metso
-
[34]
Schmidt, H.-C
J. Schmidt, H.-C. Wang, T. F. T. Cerqueira, S. Botti, and M. A. L. Marques, A dataset of 175k stable and metastable materials calculated with the PBEsol and SCAN functionals, Scientific Data9, 64 (2022)
2022
-
[35]
Zagorac, H
D. Zagorac, H. M¨ uller, S. Ruehl, J. Zagorac, and S. Rehme, Recent developments in the Inorganic Crystal Structure Database: Theoretical crystal structure data and related features, Journal of Applied Crystallography 52, 918 (2019)
2019
-
[36]
Crovetto, T
A. Crovetto, T. Unold, and A. Zakutayev, Is Cu3–xP a Semiconductor, a Metal, or a Semimetal?, Chemistry of Materials35, 1259 (2023)
2023
-
[37]
Crovetto, D
A. Crovetto, D. Kojda, F. Yi, K. N. Heinselman, D. A. LaVan, K. Habicht, T. Unold, and A. Zakutayev, Crys- tallize It before It Diffuses: Kinetic Stabilization of Thin- Film Phosphorus-Rich Semiconductor CuP2, Journal of the American Chemical Society144, 13334 (2022)
2022
-
[38]
M. H. Møuller and W. Jeitschko, Darstellung, Eigen- schaften und Kristallstruktur von Cu2P7 und Struk- turverfeinerungen von CuP2 und AgP2, Zeitschrift f¨ ur anorganische und allgemeine Chemie491, 225 (1982)
1982
-
[39]
J. He, S. H. Lee, F. Naccarato, G. Brunin, R. Zu, Y. Wang, L. Miao, H. Wang, N. Alem, G. Hautier, G.-M. Rignanese, Z. Mao, and V. Gopalan, SnP2S6: A Promising Infrared Nonlinear Optical Crystal with Strong Nonresonant Second Harmonic Generation and Phase-Matchability, ACS Photonics9, 1724 (2022)
2022
-
[40]
Dittmar and H
G. Dittmar and H. Sch¨ afer, Die Struktur des Di-Zinn- Hexathiohypodiphosphats Sn2P2S6 / The Crystal Struc- ture of Sn2P2Se, Zeitschrift f¨ ur Naturforschung B29, 312 (1974)
1974
-
[41]
W. Sun, S. T. Dacek, S. P. Ong, G. Hautier, A. Jain, W. D. Richards, A. C. Gamst, K. A. Persson, and G. Ceder, The thermodynamic scale of inorganic crys- talline metastability, Science Advances2, e1600225 (2016)
2016
-
[42]
Diehl and C.-D
R. Diehl and C.-D. Carpentier, The crystal structure of chromium thiophosphate, CrPS4, Acta Crystallograph- ica Section B: Structural Crystallography and Crystal Chemistry33, 1399 (1977)
1977
-
[43]
W. F. Kuhs, R. Nitsche, and K. Scheunemann, The ar- gyrodites — A new family of tetrahedrally close-packed structures, Materials Research Bulletin14, 241 (1979)
1979
-
[44]
Ouvrard, R
G. Ouvrard, R. Brec, and J. Rouxel, Structural determi- nation of some MPS3 layered phases (M = Mn, Fe, Co, Ni and Cd), Materials Research Bulletin20, 1181 (1985)
1985
-
[45]
Scott, M
B. Scott, M. Pressprich, R. D. Willet, and D. A. Cleary, High temperature crystal structure and DSC of Sn2P2S6, Journal of Solid State Chemistry96, 294 (1992)
1992
-
[46]
Z. Wang, R. D. Willett, R. A. Laitinen, and D. A. Cleary, Synthesis and Crystal Structure of SnP2S6, Chemistry of Materials7, 856 (1995)
1995
-
[47]
Buck and C
P. Buck and C. D. Carpentier, The crystal structure of gallium thiophosphate, GaPS4, Acta Crystallographica Section B Structural Crystallography and Crystal Chem- istry29, 1864 (1973)
1973
-
[48]
Homma, M
K. Homma, M. Yonemura, T. Kobayashi, M. Nagao, M. Hirayama, and R. Kanno, Crystal structure and phase transitions of the lithium ionic conductor Li3PS4, Solid State Ionics182, 53 (2011)
2011
-
[49]
K. Kaup, L. Zhou, A. Huq, and L. F. Nazar, Impact of the Li substructure on the diffusion pathways in alpha and beta Li3PS4: An in situ high temperature neutron diffraction study, Journal of Materials Chemistry A8, 12446 (2020)
2020
-
[50]
Scholz, C
T. Scholz, C. Schneider, R. Eger, V. Duppel, I. Moudrakovski, A. Schulz, J. Nuss, and B. V. Lotsch, Phase formation through synthetic control: Polymor- phism in the sodium-ion solid electrolyte Na4P2S6, Jour- nal of Materials Chemistry A9, 8692 (2021)
2021
-
[51]
Simon, H
A. Simon, H. Hahn, and K. Peters, Darstellung und Auf- bau von HfP2S6 / Preparation of HfP2 S6, Zeitschrift f¨ ur Naturforschung B40, 730 (1985)
1985
-
[52]
P. E. Bl¨ ochl, Projector augmented-wave method, Physi- cal Review B50, 17953 (1994)
1994
-
[53]
J. P. Perdew, A. Ruzsinszky, G. I. Csonka, O. A. Vy- drov, G. E. Scuseria, L. A. Constantin, X. Zhou, and K. Burke, Restoring the Density-Gradient Expansion for Exchange in Solids and Surfaces, Physical Review Letters 100, 136406 (2008)
2008
-
[54]
Kresse and J
G. Kresse and J. Hafner, Ab initio molecular dynamics for liquid metals, Physical Review B47, 558 (1993)
1993
-
[55]
Kresse and J
G. Kresse and J. Hafner, Ab initio molecular-dynamics simulation of the liquid-metal–amorphous-semiconductor transition in germanium, Physical Review B49, 14251 13 (1994)
1994
-
[56]
Kresse and J
G. Kresse and J. Furthm¨ uller, Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set, Computational Materials Science 6, 15 (1996)
1996
-
[57]
Kresse and J
G. Kresse and J. Furthm¨ uller, Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set, Physical Review B54, 11169 (1996)
1996
-
[58]
Kresse and D
G. Kresse and D. Joubert, From ultrasoft pseudopoten- tials to the projector augmented-wave method, Physical Review B59, 1758 (1999)
1999
-
[59]
C. J. Bartel, Review of computational approaches to predict the thermodynamic stability of inorganic solids, Journal of Materials Science57, 10475 (2022)
2022
-
[60]
A. Wang, R. Kingsbury, M. McDermott, M. Horton, A. Jain, S. P. Ong, S. Dwaraknath, and K. A. Persson, A framework for quantifying uncertainty in DFT energy corrections, Scientific Reports11, 15496 (2021)
2021
-
[61]
Zhang, D
Y. Zhang, D. A. Kitchaev, J. Yang, T. Chen, S. T. Dacek, R. A. Sarmiento-P´ erez, M. A. L. Marques, H. Peng, G. Ceder, J. P. Perdew, and J. Sun, Efficient first- principles prediction of solid stability: Towards chemical accuracy, npj Computational Materials4, 9 (2018)
2018
-
[62]
Pandey and K
M. Pandey and K. W. Jacobsen, Heats of formation of solids with error estimation: The mBEEF functional with and without fitted reference energies, Physical Re- view B91, 235201 (2015)
2015
-
[63]
Lany, Semiconductor thermochemistry in density func- tional calculations, Physical Review B78, 245207 (2008)
S. Lany, Semiconductor thermochemistry in density func- tional calculations, Physical Review B78, 245207 (2008)
2008
-
[64]
Togo, First-principles Phonon Calculations with Phonopy and Phono3py, Journal of the Physical Society of Japan92, 012001 (2023)
A. Togo, First-principles Phonon Calculations with Phonopy and Phono3py, Journal of the Physical Society of Japan92, 012001 (2023)
2023
-
[65]
Chas. G. Maier and K. K. Kelley, An equation for the rep- resentation of high-temperature heat content data, Jour- nal of the American Chemical Society54, 3243 (1932)
1932
-
[66]
I. Mills, International Union of Pure and Applied Chem- istry, and International Union of Pure and Applied Chemistry, eds.,Quantities, Units, and Symbols in Phys- ical Chemistry, 2nd ed. (Blackwell Scientific Publications ; CRC Press [distributor], Oxford ; Boston : Boca Raton, Fla, 1993)
1993
-
[67]
M. B. Ewing, T. H. Lilley, G. M. Olofsson, M. T. R¨ atzsch, and G. Somsen, Iupac: A report of IUPAC commission 1.2 on thermodynamics: Standard quantities in chemi- cal thermodynamics. Fugacities, activities, and equilib- rium constants for pure and mixed phases, The Journal of Chemical Thermodynamics27, 1 (1995)
1995
-
[68]
Epifano and D
E. Epifano and D. Monceau, Ellingham diagram: A new look at an old tool, Corrosion Science217, 111113 (2023)
2023
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.