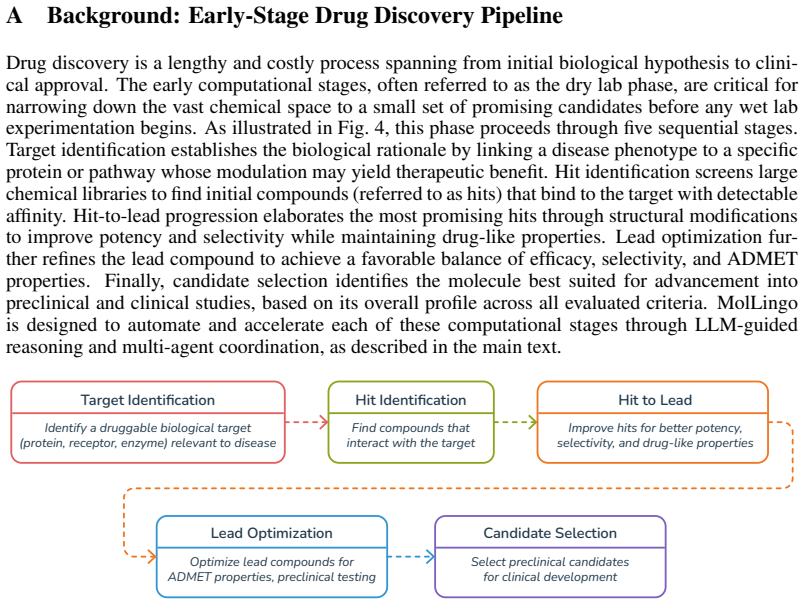
MolLingo: Molecule-Native Representations for LLM-Powered Scientific Agents
Pith reviewed 2026-06-29 12:44 UTC · model grok-4.3
The pith
LLMs become capable molecular design assistants when guided by chemically meaningful fragment representations and docking context.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
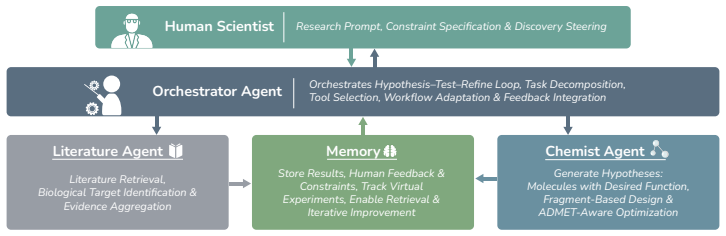
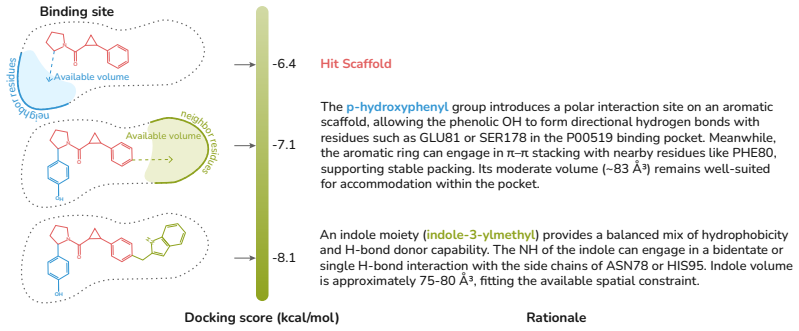
MolLingo coordinates agents through shared memory and equips them with domain tools, using BRICS-based Fragment Enumeration to represent molecules as block-based SMILES with names so that LLMs can perform block-level reasoning and editing, while grounding optimization in docking-derived residue-level protein context to improve binding.
What carries the argument
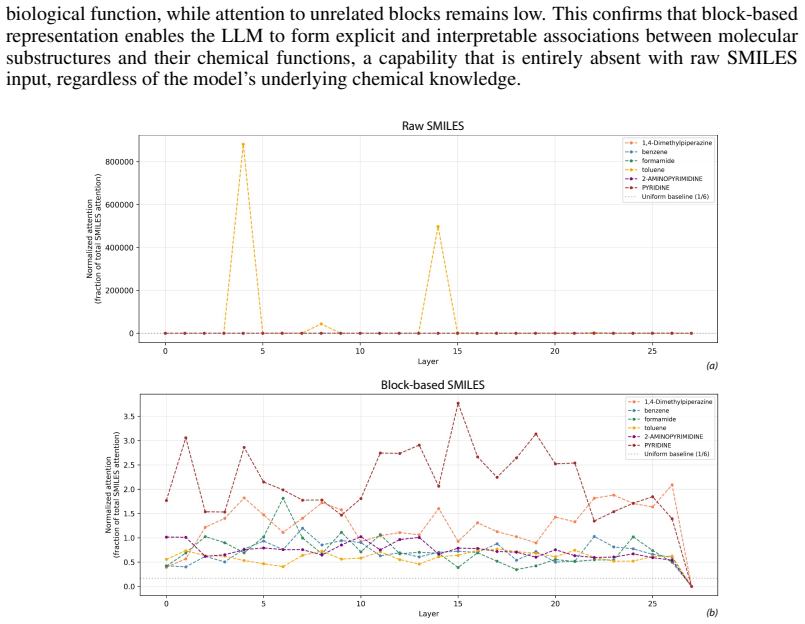
BRICS-based Fragment Enumeration (BFE), which decomposes molecules into chemically meaningful building blocks represented as block-based SMILES paired with common chemical names to bridge structure and LLM semantic space.
If this is right
- The system achieves a fourfold docking score improvement over GPT-5.4 on the same underlying model.
- It delivers consistent drug property optimization gains across multiple LLM backbones.
- It reaches state-of-the-art results on TOMG-Bench, surpassing both frontier LLMs and the RL-based RePO method.
- Multi-agent coordination with shared memory enables evidence-driven reasoning across the molecular design pipeline.
Where Pith is reading between the lines
- BFE-style fragment representations could be tested on materials or catalyst design tasks that also require synthesis-aware editing.
- The shared-memory multi-agent pattern might reduce compounding errors in other long scientific workflows such as reaction planning.
- Adding experimental assay feedback loops to the docking context would test whether the design gains translate to measured activity.
- The block-level editing might allow LLMs to incorporate synthetic accessibility constraints more directly than atom-level methods.
Load-bearing premise
The performance gains come primarily from the BFE representation and multi-agent coordination with docking context rather than from unstated prompt details or benchmark choices.
What would settle it
An ablation study that disables the BFE module, keeps all other components fixed, and re-runs the four benchmarks to check whether scores drop to levels seen with raw SMILES or unguided LLMs.
Figures


read the original abstract
We present MolLingo, a multi-agent system that emulates the reasoning process of a chemist to automate molecular design. Existing LLM-based approaches either operate as standalone generative models without access to external tools or lack the multi-agent coordination and shared memory needed for iterative, evidence-driven reasoning across the molecular design pipeline. MolLingo addresses this by coordinating a Literature Agent, a Chemist Agent, and an Orchestrator through a shared memory module, with each agent equipped with domain-specific tools. To enable effective molecular reasoning, we introduce BRICS-based Fragment Enumeration (BFE), a synthesis-aware molecular fragmentation method that decomposes molecules into chemically meaningful building blocks represented as block-based SMILES paired with common chemical names. This representation bridges molecular structure and LLM semantic space, enabling block-level reasoning and editing that is difficult with raw SMILES alone. As a case study in early-stage therapeutic design, MolLingo further grounds the Chemist Agent's reasoning in binding site geometry and residue-level protein context derived from molecular docking to optimize molecules for stronger target binding. Across four benchmarks, MolLingo consistently outperforms frontier LLMs and specialized baselines, including a fourfold docking score improvement over GPT-5.4 despite using the same underlying model, consistent drug property optimization gains across multiple LLM backbones, and state-of-the-art results on TOMG-Bench, surpassing both frontier LLMs and the RL-based optimization method RePO. Our results suggest that LLMs are already capable molecular design assistants when guided through chemically meaningful representations and biologically grounded structural context. Code is available at: https://anonymous.4open.science/status/MolLingo-7450.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript presents MolLingo, a multi-agent LLM system for molecular design comprising a Literature Agent, Chemist Agent, and Orchestrator coordinated via shared memory, each equipped with domain tools. It introduces BRICS-based Fragment Enumeration (BFE) as a synthesis-aware block-level molecular representation (block SMILES paired with chemical names) and grounds reasoning in docking-derived binding-site geometry. The central claim is that this setup yields consistent benchmark gains over frontier LLMs and specialized baselines, including a fourfold docking-score improvement over GPT-5.4 on the same backbone and SOTA results on TOMG-Bench.
Significance. If the performance gains can be isolated to the BFE representation and multi-agent loop through controlled experiments, the work would provide concrete evidence that chemically meaningful, block-level representations plus biologically grounded context enable LLMs to function as iterative molecular design assistants.
major comments (2)
- [Abstract and §4] Abstract and §4 (Experimental Results): the reported fourfold docking improvement and cross-benchmark gains are presented without any ablation that holds the base LLM, prompt budget, and tool access fixed while removing either the BFE representation or the three-agent division; this directly undermines the claim that gains arise from the proposed mechanisms rather than unstated implementation choices.
- [§4] §4: no experimental protocol, number of independent runs, error bars, or statistical tests are supplied for the docking or TOMG-Bench results, so the quantitative claims cannot be reproduced or compared to the skeptic baseline of equally-prompted single-agent or tool-augmented controls.
minor comments (2)
- The anonymous code link should be replaced with a permanent repository upon acceptance to support reproducibility.
- [§3] Notation for BFE blocks (e.g., how SMILES fragments are paired with names) is introduced in §3 but lacks a small illustrative table or figure showing an example decomposition.
Simulated Author's Rebuttal
Thank you for the constructive feedback. We address the major comments below and will revise the manuscript to strengthen the experimental section.
read point-by-point responses
-
Referee: [Abstract and §4] Abstract and §4 (Experimental Results): the reported fourfold docking improvement and cross-benchmark gains are presented without any ablation that holds the base LLM, prompt budget, and tool access fixed while removing either the BFE representation or the three-agent division; this directly undermines the claim that gains arise from the proposed mechanisms rather than unstated implementation choices.
Authors: We agree that the manuscript does not contain the requested controlled ablations that fix the base LLM, prompt budget, and tool access while removing BFE or the three-agent structure. The current text only notes performance 'despite using the same underlying model' without isolating the other factors. In revision we will add these ablations to allow direct attribution of gains to the proposed components. revision: yes
-
Referee: [§4] §4: no experimental protocol, number of independent runs, error bars, or statistical tests are supplied for the docking or TOMG-Bench results, so the quantitative claims cannot be reproduced or compared to the skeptic baseline of equally-prompted single-agent or tool-augmented controls.
Authors: We acknowledge that §4 currently omits the experimental protocol details, number of independent runs, error bars, and statistical tests. The revised version will expand this section with a full protocol description, the number of runs performed, means with standard deviations, and appropriate statistical comparisons to enable reproduction and fair evaluation against single-agent baselines. revision: yes
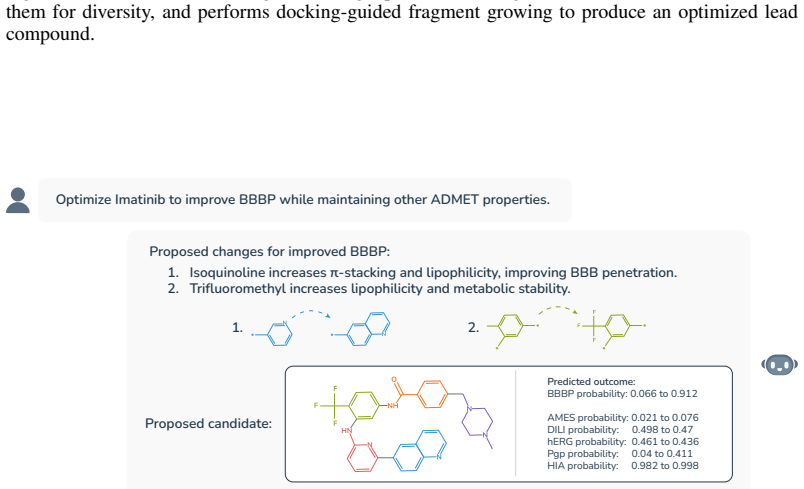
Circularity Check
No significant circularity; empirical claims rest on external benchmarks.
full rationale
The paper introduces BFE as a fragmentation method and a multi-agent architecture, then reports benchmark performance (e.g., docking scores, TOMG-Bench) against frontier LLMs and baselines. No equations, fitted parameters, or self-citations appear in the provided text that reduce any claimed result to a tautology or prior input by construction. All load-bearing evidence consists of comparative evaluations on independent test sets, satisfying the self-contained criterion.
Axiom & Free-Parameter Ledger
axioms (1)
- domain assumption Molecules can be decomposed into chemically meaningful fragments using BRICS rules that preserve synthetic accessibility.
Reference graph
Works this paper leans on
-
[1]
Josh Achiam, Steven Adler, Sandhini Agarwal, Lama Ahmad, Ilge Akkaya, Florencia Leoni Aleman, Diogo Almeida, Janko Altenschmidt, Sam Altman, Shyamal Anadkat, et al. Gpt-4 technical report.arXiv preprint arXiv:2303.08774, 2023
work page internal anchor Pith review Pith/arXiv arXiv 2023
-
[2]
Claude 3 model card
Anthropic. Claude 3 model card. Technical report, Anthropic, 2024. URLhttps://www. anthropic.com
2024
-
[3]
The properties of known drugs
Guy W Bemis and Mark A Murcko. The properties of known drugs. 1. molecular frameworks. Journal of medicinal chemistry, 39(15):2887–2893, 1996
1996
-
[4]
The protein data bank.Nucleic acids research, 28(1):235–242, 2000
Helen M Berman, John Westbrook, Zukang Feng, Gary Gilliland, Talapady N Bhat, Helge Weissig, Ilya N Shindyalov, and Philip E Bourne. The protein data bank.Nucleic acids research, 28(1):235–242, 2000
2000
-
[5]
Computational fragment-based drug design: current trends, strategies, and applications.The AAPS journal, 20(3):59, 2018
Yuemin Bian and Xiang-Qun Xie. Computational fragment-based drug design: current trends, strategies, and applications.The AAPS journal, 20(3):59, 2018
2018
-
[6]
Quantifying the chemical beauty of drugs.Nature chemistry, 4(2):90–98, 2012
G Richard Bickerton, Gaia V Paolini, J´er´emy Besnard, Sorel Muresan, and Andrew L Hopkins. Quantifying the chemical beauty of drugs.Nature chemistry, 4(2):90–98, 2012
2012
-
[7]
Autonomous chemical research with large language models.Nature, 624(7992):570–578, 2023
Daniil A Boiko, Robert MacKnight, Ben Kline, and Gabe Gomes. Autonomous chemical research with large language models.Nature, 624(7992):570–578, 2023
2023
-
[8]
van der waals volumes and radii.The Journal of physical chemistry, 68(3): 441–451, 1964
A van Bondi. van der waals volumes and radii.The Journal of physical chemistry, 68(3): 441–451, 1964
1964
-
[9]
ChemCrow: Augmenting large-language models with chemistry tools
Andres M Bran, Sam Cox, Andrew D White, and Philippe Schwaller. Chemcrow: Augmenting large-language models with chemistry tools.arXiv preprint arXiv:2304.05376, 2023
work page internal anchor Pith review Pith/arXiv arXiv 2023
-
[10]
Darko Butina. Unsupervised data base clustering based on daylight’s fingerprint and tanimoto similarity: A fast and automated way to cluster small and large data sets.Journal of Chemical Information and Computer Sciences, 39(4):747–750, 1999
1999
-
[11]
A’rule of three’for fragment- based lead discovery?Drug discovery today, 8(19):876–877, 2003
Miles Congreve, Robin Carr, Chris Murray, and Harren Jhoti. A’rule of three’for fragment- based lead discovery?Drug discovery today, 8(19):876–877, 2003
2003
-
[12]
Gemini model technical report / model card
Google DeepMind. Gemini model technical report / model card. Technical report, Google DeepMind, 2024. URLhttps://deepmind.google
2024
-
[13]
On the art of compiling and using’drug-like’chemical fragment spaces.ChemMedChem, 3(10):1503, 2008
Jorg Degen, Christof Wegscheid-Gerlach, Andrea Zaliani, and Matthias Rarey. On the art of compiling and using’drug-like’chemical fragment spaces.ChemMedChem, 3(10):1503, 2008
2008
-
[14]
Carl Edwards, Chi Han, Gawon Lee, Thao Nguyen, Bowen Jin, Chetan Kumar Prasad, Sara Szymkuc, Bartosz A Grzybowski, Ying Diao, Jiawei Han, et al. mclm: A function-infused and synthesis-friendly modular chemical language model.arXiv preprint arXiv:2505.12565, 2025
-
[15]
Estimation of synthetic accessibility score of drug-like molecules based on molecular complexity and fragment contributions.Journal of cheminfor- matics, 1(1):8, 2009
Peter Ertl and Ansgar Schuffenhauer. Estimation of synthetic accessibility score of drug-like molecules based on molecular complexity and fragment contributions.Journal of cheminfor- matics, 1(1):8, 2009
2009
-
[16]
Yao Fehlis, Charles Crain, Aidan Jensen, Michael Watson, James Juhasz, Paul Mandel, Betty Liu, Shawn Mahon, Daren Wilson, Nick Lynch-Jonely, et al. Accelerating drug discovery through agentic ai: A multi-agent approach to laboratory automation in the dmta cycle.arXiv preprint arXiv:2507.09023, 2025
-
[17]
Withdrawn 2.0—update on withdrawn drugs with pharmacovigilance data.Nucleic Acids Research, 52(D1):D1503–D1507, 2024
Kathleen Gallo, Andrean Goede, Oliver-Andreas Eckert, Bjoern-Oliver Gohlke, and Robert Preissner. Withdrawn 2.0—update on withdrawn drugs with pharmacovigilance data.Nucleic Acids Research, 52(D1):D1503–D1507, 2024
2024
-
[18]
Drugclip: Contrastive protein-molecule representation learning for virtual screening.Advances in Neural Information Processing Systems, 36:44595–44614, 2023
Bowen Gao, Bo Qiang, Haichuan Tan, Yinjun Jia, Minsi Ren, Minsi Lu, Jingjing Liu, Wei- Ying Ma, and Yanyan Lan. Drugclip: Contrastive protein-molecule representation learning for virtual screening.Advances in Neural Information Processing Systems, 36:44595–44614, 2023. 11
2023
-
[19]
Bowen Gao, Yanwen Huang, Yiqiao Liu, Wenxuan Xie, Wei-Ying Ma, Ya-Qin Zhang, and Yanyan Lan. Pharmagents: Building a virtual pharma with large language model agents.arXiv preprint arXiv:2503.22164, 2025
-
[20]
Chembl: a large-scale bioactivity database for drug discovery.Nucleic acids research, 40(D1):D1100– D1107, 2012
Anna Gaulton, Louisa J Bellis, A Patricia Bento, Jon Chambers, Mark Davies, Anne Hersey, Yvonne Light, Shaun McGlinchey, David Michalovich, Bissan Al-Lazikani, et al. Chembl: a large-scale bioactivity database for drug discovery.Nucleic acids research, 40(D1):D1100– D1107, 2012
2012
-
[21]
What can large language models do in chemistry? a comprehensive benchmark on eight tasks.Advances in neural information processing systems, 36:59662– 59688, 2023
Taicheng Guo, Bozhao Nan, Zhenwen Liang, Zhichun Guo, Nitesh Chawla, Olaf Wiest, Xi- angliang Zhang, et al. What can large language models do in chemistry? a comprehensive benchmark on eight tasks.Advances in neural information processing systems, 36:59662– 59688, 2023
2023
-
[22]
Kexin Huang, Tianfan Fu, Wenhao Gao, Yue Zhao, Yusuf Roohani, Jure Leskovec, Con- nor W Coley, Cao Xiao, Jimeng Sun, and Marinka Zitnik. Therapeutics data commons: Machine learning datasets and tasks for drug discovery and development.arXiv preprint arXiv:2102.09548, 2021
-
[23]
Junkai Ji, Zhangfan Yang, Dong Xu, Ruibin Bai, Jianqiang Li, Tingjun Hou, and Zexuan Zhu. Toward closed-loop molecular discovery via language model, property alignment and strategic search.arXiv preprint arXiv:2512.09566, 2025
-
[24]
Highly accurate protein structure prediction with alphafold.nature, 596(7873):583–589, 2021
John Jumper, Richard Evans, Alexander Pritzel, Tim Green, Michael Figurnov, Olaf Ron- neberger, Kathryn Tunyasuvunakool, Russ Bates, Augustin ˇZ´ıdek, Anna Potapenko, et al. Highly accurate protein structure prediction with alphafold.nature, 596(7873):583–589, 2021
2021
-
[25]
Fragberta: Exploring fragment-based molecular representation learning with safe
Neerav Kaushal and Ajay MNV Penmatsa. Fragberta: Exploring fragment-based molecular representation learning with safe. 2026
2026
-
[26]
Patrick S. H. Lewis, Ethan Perez, Aleksandra Piktus, Fabio Petroni, Vladimir Karpukhin, Naman Goyal, Heinrich K ¨uttler, Mike Lewis, Wen-tau Yih, Tim Rockt ¨aschel, Sebastian Riedel, and Douwe Kiela. Retrieval-augmented generation for knowledge-intensive NLP tasks. In Hugo Larochelle, Marc’Aurelio Ranzato, Raia Hadsell, Maria-Florina Balcan, and Hsuan-Tie...
2020
-
[27]
Speak-to-Structure: Evaluating LLMs in Open-domain Natural Language-Driven Molecule Generation
Jiatong Li, Junxian Li, Yunqing Liu, Dongzhan Zhou, and Qing Li. Tomg-bench: Evaluating llms on text-based open molecule generation.arXiv preprint arXiv:2412.14642, 2024
work page internal anchor Pith review Pith/arXiv arXiv 2024
-
[28]
Xuan Li, Zhanke Zhou, Zongze Li, Jiangchao Yao, Yu Rong, Lu Zhang, and Bo Han. Reference-guided policy optimization for molecular optimization via llm reasoning.arXiv preprint arXiv:2603.05900, 2026
-
[29]
Yizhan Li, Florence Cloutier, Sifan Wu, Ali Parviz, Boris Knyazev, Yan Zhang, Glen Berseth, and Bang Liu. Mˆ 4olgen: Multi-agent, multi-stage molecular generation under precise multi- property constraints.arXiv preprint arXiv:2601.10131, 2026
-
[30]
Thomas J Lynch, Daphne W Bell, Raffaella Sordella, Sarada Gurubhagavatula, Ross A Oki- moto, Brian W Brannigan, Patricia L Harris, Sara M Haserlat, Jeffrey G Supko, Frank G Haluska, et al. Activating mutations in the epidermal growth factor receptor underlying re- sponsiveness of non–small-cell lung cancer to gefitinib.New England Journal of Medicine, 350...
2004
-
[31]
FARM: Enhancing Molecular Representations with Functional Group Awareness
Thao Nguyen, Kuan-Hao Huang, Ge Liu, Martin D Burke, Ying Diao, and Heng Ji. Farm: Functional group-aware representations for small molecules.arXiv preprint arXiv:2410.02082, 2024
work page internal anchor Pith review Pith/arXiv arXiv 2024
-
[32]
Team Qwen. Qwen2 technical report.arXiv preprint arXiv:2407.10671, 2024. URLhttps: //arxiv.org/abs/2407.10671. 12
work page internal anchor Pith review Pith/arXiv arXiv 2024
-
[33]
Computational approaches streamlining drug discovery.Nature, 616(7958):673–685, 2023
Anastasiia V Sadybekov and Vsevolod Katritch. Computational approaches streamlining drug discovery.Nature, 616(7958):673–685, 2023
2023
-
[34]
FragmentNet: Adaptive Graph Fragmentation for Graph-to-Sequence Molecular Representation Learning
Ankur Samanta, Rohan Gupta, Aditi Misra, Christian McIntosh Clarke, and Jayakumar Ra- jadas. Fragmentnet: Adaptive graph fragmentation for graph-to-sequence molecular represen- tation learning.arXiv preprint arXiv:2502.01184, 2025
work page internal anchor Pith review Pith/arXiv arXiv 2025
-
[35]
Graphbpe: Molecular graphs meet byte-pair encoding
Yuchen Shen and Barnab ´as P ´oczos. Graphbpe: Molecular graphs meet byte-pair encoding. arXiv preprint arXiv:2407.19039, 2024
-
[36]
Madd: Multi-agent drug discovery orchestra
Gleb V Solovev, Alina B Zhidkovskaya, Anastasia Orlova, Nina Gubina, Anastasia Vepreva, Rodion Golovinskii, Ilya Tonkii, Ivan Dubrovsky, Ivan Gurev, Dmitry Gilemkhanov, et al. Madd: Multi-agent drug discovery orchestra. InFindings of the Association for Computational Linguistics: EMNLP 2025, pages 6956–6998, 2025
2025
-
[37]
Chemical fragments as foundations for understanding target space and activity prediction.Journal of medicinal chemistry, 51(9):2689–2700, 2008
Jeffrey J Sutherland, Richard E Higgs, Ian Watson, and Michal Vieth. Chemical fragments as foundations for understanding target space and activity prediction.Journal of medicinal chemistry, 51(9):2689–2700, 2008
2008
-
[38]
Elementary mathematical theory of classification and prediction
Taffee T Tanimoto. Elementary mathematical theory of classification and prediction. 1958
1958
-
[39]
Oleg Trott and Arthur J Olson. Autodock vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading.Journal of computa- tional chemistry, 31(2):455–461, 2010
2010
-
[40]
Reconstruction of lossless molecular representations from fingerprints.Journal of cheminformatics, 15(1):26, 2023
Umit V Ucak, Islambek Ashyrmamatov, and Juyong Lee. Reconstruction of lossless molecular representations from fingerprints.Journal of cheminformatics, 15(1):26, 2023
2023
-
[41]
Target-specific de novo design of drug candidate molecules with graph- transformer-based generative adversarial networks.Nature Machine Intelligence, 7(9):1524– 1540, 2025
Atabey ¨Unl¨u, Elif C ¸ evrim, Melih G¨okay Yi˘git, Ahmet Sarıg¨un, Hayriye C ¸ elikbilek, Osman Bayram, Deniz Cansen Kahraman, Abdurrahman Ol ˘gac ¸, Ahmet Sureyya Rifaioglu, Erden Bano˘glu, et al. Target-specific de novo design of drug candidate molecules with graph- transformer-based generative adversarial networks.Nature Machine Intelligence, 7(9):152...
2025
-
[42]
Remol: Llm-guided molecular optimization with reinforcement learning
Ziqing Wang and Kaize Ding. Remol: Llm-guided molecular optimization with reinforcement learning. 2018
2018
-
[43]
Smiles, a chemical language and information system
David Weininger. Smiles, a chemical language and information system. 1. introduction to methodology and encoding rules.Journal of chemical information and computer sciences, 28 (1):31–36, 1988
1988
-
[44]
React: Synergizing reasoning and acting in language models
Shunyu Yao, Jeffrey Zhao, Dian Yu, Nan Du, Izhak Shafran, Karthik R Narasimhan, and Yuan Cao. React: Synergizing reasoning and acting in language models. InThe eleventh international conference on learning representations, 2022
2022
-
[45]
Jie Yue, Bingxin Peng, Yu Chen, Jieyu Jin, Xinda Zhao, Chao Shen, Xiangyang Ji, Chang-Yu Hsieh, Jianfei Song, Tingjun Hou, et al. Unlocking comprehensive molecular design across all scenarios with large language model and unordered chemical language.Chemical Science, 15 (34):13727–13740, 2024. 13 A Background: Early-Stage Drug Discovery Pipeline Drug disc...
-
[46]
Isoquinoline increases π-stacking and lipophilicity, improving BBB penetration
-
[47]
Trifluoromethyl increases lipophilicity and metabolic stability
-
[48]
2. Proposed candidate: Predicted outcome: BBBP probability: 0.066 to 0.912 AMES probability: 0.021 to 0.076 DILI probability: 0.498 to 0.47 hERG probability: 0.461 to 0.436 Pgp probability: 0.04 to 0.411 HIA probability: 0.982 to 0.998 Figure 8: Lead optimization: the Chemist Agent iteratively refines a lead molecule through block- level modifications gui...
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.