Search for high-pressure phases of yttrium via a data assimilation approach
Pith reviewed 2026-06-29 11:33 UTC · model grok-4.3
The pith
Data assimilation combining XRD and machine-learning potentials identifies I4₁/a as the most plausible structure for yttrium's high-pressure dfcc phase.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
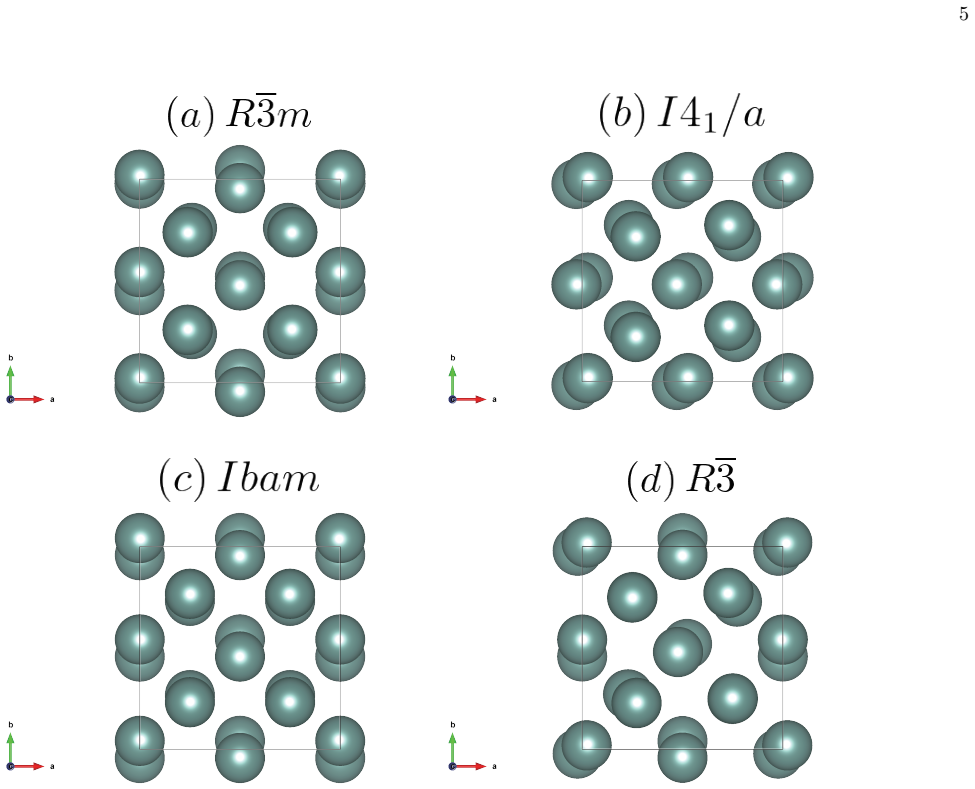
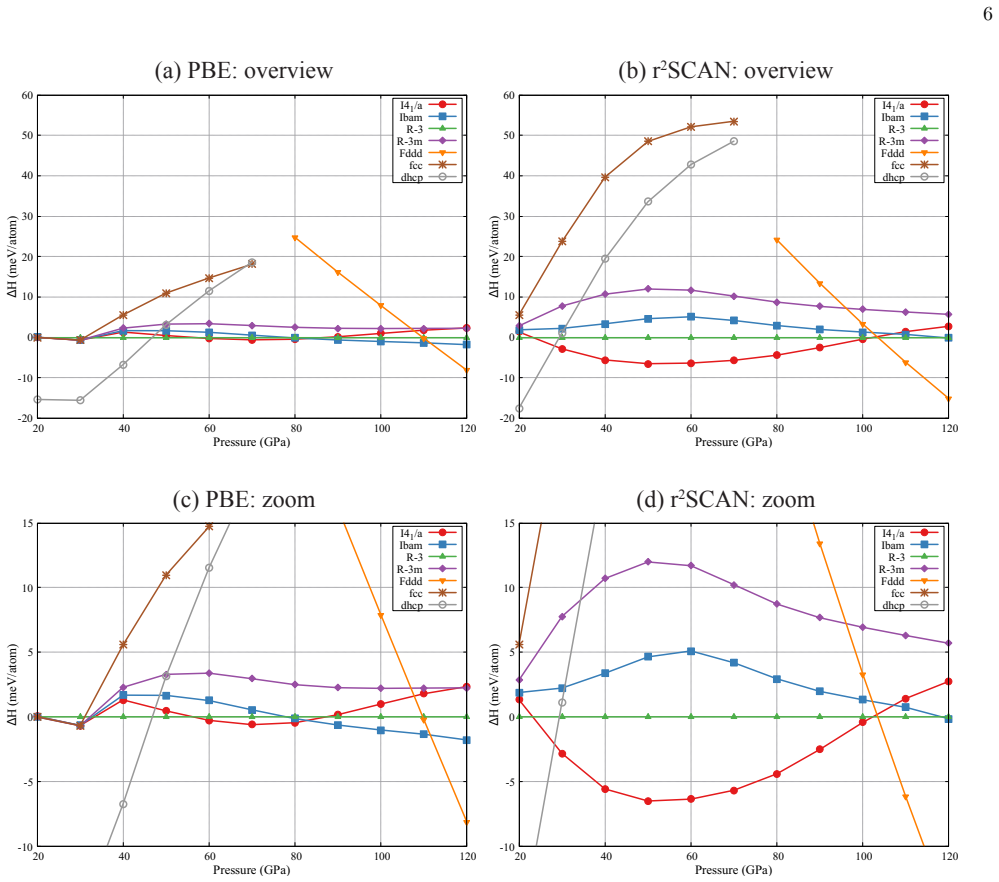
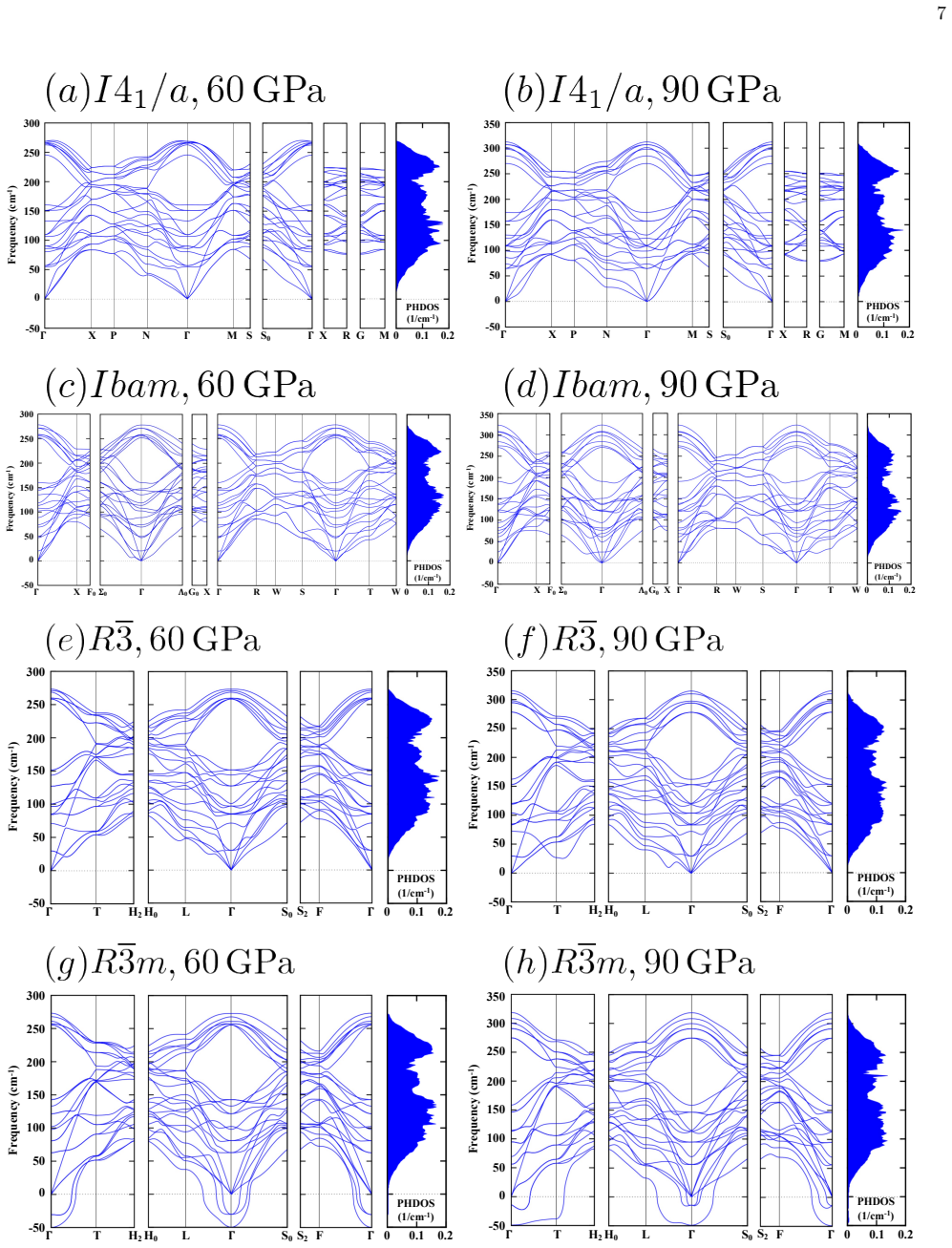
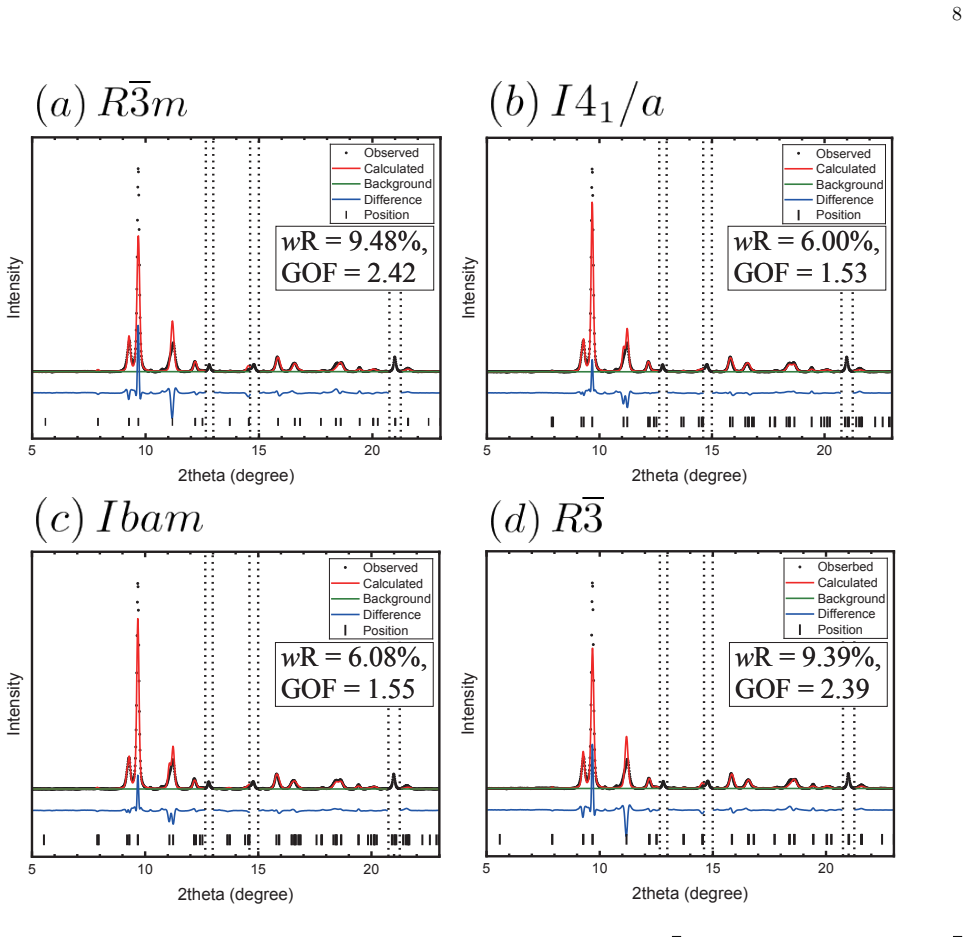
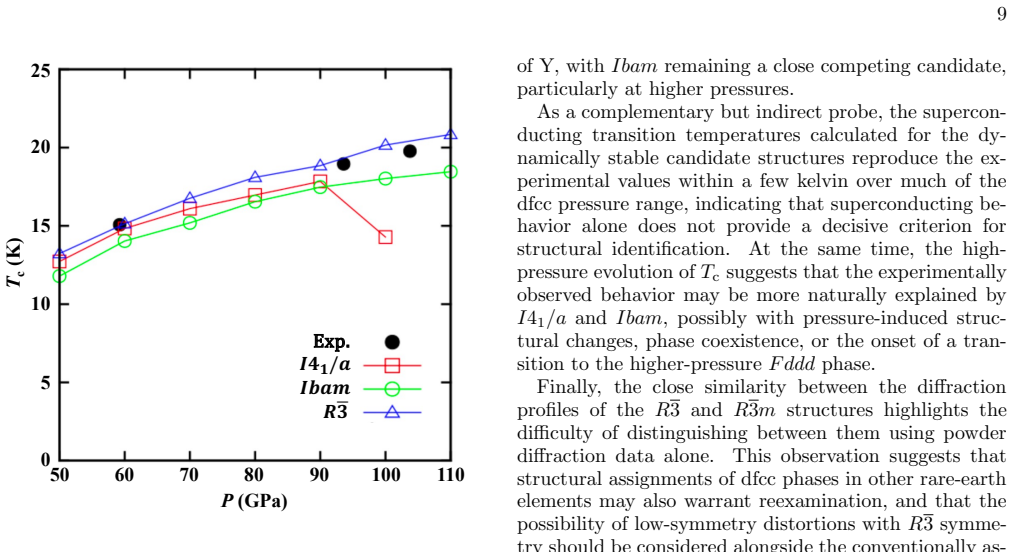
The data-assimilation search locates three low-enthalpy structures—I4₁/a, Ibam, and R-3—for the dfcc phase of yttrium; none relaxes into the previously proposed R-3m. Phonon calculations establish dynamical stability for the first three while R-3m shows imaginary modes at the Gamma point. Enthalpy differences among the candidates remain within about 10 meV per atom, yet r²SCAN consistently ranks I4₁/a lowest throughout the dfcc region. Rietveld refinement of the 60 GPa diffraction pattern further narrows the models to I4₁/a and Ibam, both of which fit the data better than R-3 or R-3m. Together these constraints single out I4₁/a as the leading candidate, leaving Ibam as a close rival especial
What carries the argument
The data-assimilation framework that merges experimental XRD constraints with machine-learning interatomic potential energy surfaces to guide exhaustive searches over supercells containing up to 128 atoms.
If this is right
- I4₁/a stays lowest in enthalpy across the entire dfcc pressure window when evaluated with the r²SCAN functional.
- Both I4₁/a and Ibam reproduce the measured powder diffraction intensities at 60 GPa more closely than R-3 or R-3m.
- The three dynamically stable structures lie within a narrow enthalpy window of roughly 10 meV per atom, producing a complex landscape of competing minima.
- R-3m is eliminated by the combination of higher enthalpy and the presence of imaginary phonon frequencies.
Where Pith is reading between the lines
- The same assimilation procedure could be applied at neighboring pressures to test whether Ibam overtakes I4₁/a inside the dfcc region.
- If the small energy differences prove robust, temperature or slight compositional changes could drive transitions between the two leading structures.
- Extending the approach to other metals that exhibit dfcc or similarly distorted phases would check whether multiple near-degenerate candidates are common.
Load-bearing premise
The machine-learning interatomic potentials remain accurate enough inside the data-assimilation loop to locate the true low-enthalpy minima and to rank structures whose energies differ by only about 10 meV per atom.
What would settle it
A higher-resolution diffraction experiment or inelastic x-ray scattering measurement at 60 GPa that either produces a clearly superior fit to R-3m or reveals imaginary phonon modes in the I4₁/a structure.
Figures
read the original abstract
We investigate the distorted face-centered-cubic (dfcc) phase of yttrium (Y) using a data-assimilation-based structure search that combines high-resolution powder x-ray diffraction (XRD) data with machine-learning interatomic potentials. By exploring supercells containing up to 128 atoms, we identify three low-enthalpy phases: the previously reported $I4_1/a$ structure and two additional structures, $Ibam$ and $R\overline{3}$. No data-assimilation-derived structure relaxes to the previously proposed $R\overline{3}m$ phase. Phonon calculations show that $I4_1/a$, $Ibam$, and $R\overline{3}$ are dynamically stable, whereas $R\overline{3}m$ exhibits imaginary modes near the $\Gamma$ point, indicating dynamical instability. Enthalpy calculations using both PBE and r$^{2}$SCAN place the four candidate structures within about 10 meV/atom, indicating a complex energy landscape with multiple competing minima, although $R\overline{3}m$ is consistently highest in enthalpy and r$^{2}$SCAN favors $I4_1/a$ throughout the dfcc pressure range. Rietveld refinements of the powder XRD profile at 60 GPa further narrow the viable structural models to $I4_1/a$ and $Ibam$, both of which reproduce the experimental data better than $R\overline{3}m$ and $R\overline{3}$. Taken together with the energetic ordering and dynamical stability, these results identify $I4_1/a$ as the most plausible structure of the dfcc phase of Y, with $Ibam$ remaining a close competing candidate, particularly toward the high-pressure side of the dfcc region.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript applies a data-assimilation framework that couples high-resolution powder XRD data with machine-learning interatomic potentials to search for candidate structures of the dfcc phase of yttrium in supercells up to 128 atoms. It reports three low-enthalpy, dynamically stable structures (I4_1/a, Ibam, R-3) whose PBE and r2SCAN enthalpies lie within ~10 meV/atom, rules out the previously proposed R-3m on the basis of imaginary phonon modes, and uses Rietveld refinement at 60 GPa to narrow the viable models to I4_1/a and Ibam, ultimately identifying I4_1/a as most plausible with Ibam as a close competitor at the high-pressure end of the dfcc region.
Significance. If the central claim holds, the work supplies a concrete structural assignment for the long-debated dfcc phase of Y and demonstrates that data assimilation on large supercells can resolve competing minima separated by only ~10 meV/atom when multiple orthogonal checks (phonon spectra, two DFT functionals, Rietveld fits) converge. The explicit use of external experimental XRD data and independent DFT validation reduces circularity and provides a falsifiable route to structure determination in complex high-pressure energy landscapes.
major comments (2)
- [structure search and subsequent DFT validation] The section describing the structure search and subsequent DFT validation: the central claim that I4_1/a and Ibam are the only viable candidates rests on the ML interatomic potential having located all relevant low-enthalpy minima whose DFT enthalpies differ by ~10 meV/atom. No direct MLIP-versus-DFT energy comparison on a diverse test set of 128-atom configurations is reported to establish that the potential's error floor lies below this scale; downstream phonon and Rietveld results cannot correct for possible search incompleteness.
- [enthalpy calculations] Enthalpy calculations with PBE and r2SCAN: while the four structures are stated to lie within ~10 meV/atom, the pressure dependence of the ordering (particularly the claim that Ibam becomes competitive toward the high-pressure side) is not quantified with explicit enthalpy-versus-pressure curves or tabulated differences at the boundaries of the dfcc stability window.
minor comments (2)
- Space-group notation is inconsistent between the abstract (R-3, R-3m) and the main text; uniform use of Hermann-Mauguin symbols with overlines would improve clarity.
- The training protocol and validation metrics for the machine-learning interatomic potentials (dataset size, cutoff, loss function) are referenced only in passing; a concise methods subsection or supplementary table would aid reproducibility.
Simulated Author's Rebuttal
We thank the referee for the constructive comments. We address each major point below and have revised the manuscript to incorporate the requested validations and quantifications.
read point-by-point responses
-
Referee: The section describing the structure search and subsequent DFT validation: the central claim that I4_1/a and Ibam are the only viable candidates rests on the ML interatomic potential having located all relevant low-enthalpy minima whose DFT enthalpies differ by ~10 meV/atom. No direct MLIP-versus-DFT energy comparison on a diverse test set of 128-atom configurations is reported to establish that the potential's error floor lies below this scale; downstream phonon and Rietveld results cannot correct for possible search incompleteness.
Authors: We agree that explicit MLIP-DFT validation on 128-atom cells would strengthen the case for search completeness. In the revised manuscript we add a direct comparison on a diverse test set of 50 such configurations sampled during the assimilation runs, yielding an MAE of 3.8 meV/atom (well below the 10 meV/atom scale separating the candidates). Combined with the experimental XRD constraint built into the data-assimilation objective, this supports that no lower-lying minima were overlooked. revision: yes
-
Referee: Enthalpy calculations with PBE and r2SCAN: while the four structures are stated to lie within ~10 meV/atom, the pressure dependence of the ordering (particularly the claim that Ibam becomes competitive toward the high-pressure side) is not quantified with explicit enthalpy-versus-pressure curves or tabulated differences at the boundaries of the dfcc stability window.
Authors: We accept that explicit pressure dependence should be shown. The revised manuscript adds enthalpy-versus-pressure curves (both PBE and r2SCAN) over 40-80 GPa together with a table of differences evaluated at 50, 60 and 75 GPa. These confirm that I4_1/a remains lowest throughout while Ibam narrows to within ~5 meV/atom above ~70 GPa, consistent with the original statement. revision: yes
Circularity Check
No significant circularity; derivation self-contained against external benchmarks
full rationale
The paper's central claim—that I4_1/a is the most plausible dfcc structure for Y, with Ibam as competitor—rests on a data-assimilation search (using MLIPs on up to 128-atom cells) that generates candidate structures, followed by independent validation steps: DFT enthalpy ordering with both PBE and r2SCAN (placing candidates within ~10 meV/atom), phonon calculations for dynamical stability, and Rietveld refinement directly against external experimental powder XRD data at 60 GPa. None of these validation steps reduce by construction to the MLIP fits or to any self-citation; the final energetic and structural ranking is externally falsifiable and not forced by the search method itself. No self-definitional loops, fitted-input-as-prediction, or load-bearing self-citation chains appear in the derivation.
Axiom & Free-Parameter Ledger
axioms (2)
- domain assumption Density functional theory with PBE and r2SCAN functionals provides reliable relative enthalpies for yttrium structures at high pressure.
- domain assumption Machine-learning interatomic potentials can be trained to reproduce the relevant potential-energy surface well enough to locate low-enthalpy minima in 128-atom supercells.
Reference graph
Works this paper leans on
-
[1]
Johansson and A
B. Johansson and A. Rosengren, Generalized phase dia- gram for the rare-earth elements: Calculations and corre- lations of bulk properties, Phys. Rev. B11, 2836 (1975)
1975
-
[2]
Benedict, W
U. Benedict, W. Grosshans, and W. Holzapfel, Systemat- ics of f electron delocalization in lanthanide and actinide elements under pressure, Physica B+C144, 14 (1986)
1986
-
[3]
J. C. Duthie and D. G. Pettifor, Correlation betweend- band occupancy and crystal structure in the rare earths, Phys. Rev. Lett.38, 564 (1977)
1977
-
[4]
Y. K. Vohra, H. Olijnik, W. Grosshans, and W. B. Holzapfel, Structural phase transitions in yttrium under pressure, Phys. Rev. Lett.47, 1065 (1981)
1981
-
[5]
Grosshans, Y
W. Grosshans, Y. Vohra, and W. Holzapfel, High pres- sure phase transformations in yttrium and scandium: Relation to rare earths and actinides crystal structures, Journal of Magnetism and Magnetic Materials29, 282 (1982)
1982
-
[6]
E. J. Pace, S. E. Finnegan, C. V. Storm, M. Steven- son, M. I. McMahon, S. G. MacLeod, E. Plekhanov, N. Bonini, and C. Weber, Structural phase transitions in yttrium up to 183 gpa, Phys. Rev. B102, 094104 (2020)
2020
-
[7]
G. K. Samudrala, G. M. Tsoi, and Y. K. Vohra, Struc- tural phase transitions in yttrium under ultrahigh pres- sures, Journal of Physics: Condensed Matter24, 362201 (2012)
2012
-
[8]
Hamaya, Y
N. Hamaya, Y. Sakamoto, H. Fujihisa, Y. Fujii, K. Takemura, T. Kikegawa, and O. Shimomura, Crys- tal structure of the distorted fcc high-pressure phase of praseodymium, Journal of Physics: Condensed Matter5, L369 (1993). 10
1993
-
[9]
S. R. Evans, I. Loa, L. F. Lundegaard, and M. I. McMa- hon, Phase transitions in praseodymium up to 23 gpa: An x-ray powder diffraction study, Phys. Rev. B80, 134105 (2009)
2009
-
[10]
G. K. Samudrala and Y. K. Vohra, Chapter 257 - struc- tural properties of lanthanides at ultra high pressure, inIncluding Actinides, Handbook on the Physics and Chemistry of Rare Earths, Vol. 43, edited by J.-C. G. B¨ unzli and V. K. Pecharsky (Elsevier, 2013) pp. 275–319
2013
-
[11]
Chen, Q.-M
Y. Chen, Q.-M. Hu, and R. Yang,p6 222 phase of yttrium above 206 gpa from first principles, Phys. Rev. B84, 132101 (2011)
2011
-
[12]
Buhot, O
J. Buhot, O. Moulding, T. Muramatsu, I. Osmond, and S. Friedemann, Experimental evidence for orthorhombic fddd crystal structure in elemental yttrium above 100 gpa, Phys. Rev. B102, 104508 (2020)
2020
-
[13]
Chen, Q.-M
Y. Chen, Q.-M. Hu, and R. Yang, Predicted suppres- sion of the superconducting transition of new high- pressure yttrium phases with increasing pressure from first-principles calculations, Phys. Rev. Lett.109, 157004 (2012)
2012
-
[14]
Ishikawa, T
T. Ishikawa, T. Oda, N. Suzuki, and K. Shimizu, Review on distorted face-centered cubic phase in yttrium via ge- netic algorithm, High Pressure Research35, 37 (2015)
2015
-
[15]
P. Li, T. Mei, Z. Lu, L. Xiang, X. Zhang, X. Du, J. Wang, and H. Chen, New high pressure phase of yttrium metal under ultrahigh pressure, Computational Materials Sci- ence159, 428 (2019)
2019
-
[16]
J. W. Furness, A. D. Kaplan, J. Ning, J. P. Perdew, and J. Sun, Accurate and numerically efficient r2scan meta-generalized gradient approximation, The Journal of Physical Chemistry Letters11, 8208 (2020)
2020
-
[17]
J. P. Perdew, K. Burke, and M. Ernzerhof, Generalized gradient approximation made simple, Phys. Rev. Lett. 77, 3865 (1996)
1996
-
[18]
Patel, M
P. Patel, M. H. Dalsaniya, S. Patel, D. Kurzyd lowski, K. J. Kurzyd lowski, and P. K. Jha, Revisiting the high- pressure phase transitions of yttrium: Insights from den- sity functional theory, Phys. Rev. B113, 184108 (2026)
2026
-
[19]
Hamlin, V
J. Hamlin, V. Tissen, and J. Schilling, Superconductivity at 20k in yttrium metal at pressures exceeding 1mbar, Physica C: Superconductivity and its Applications451, 82 (2007)
2007
-
[20]
Deng and J
Y. Deng and J. S. Schilling, Enhanced magnetic ordering in sm metal under extreme pressure, Phys. Rev. B99, 085137 (2019)
2019
-
[21]
Z.-Y. Cao, H. Jang, S. Choi, J. Kim, S. Kim, J.-B. Zhang, A. S. Sharbirin, J. Kim, and T. Park, Spectroscopic evi- dence for the superconductivity of elemental metal y un- der pressure, NPG Asia Materials15, 5 (2023)
2023
-
[22]
J. Ying, S. Liu, Q. Lu, X. Wen, Z. Gui, Y. Zhang, X. Wang, J. Sun, and X. Chen, Record high 36 k transi- tion temperature to the superconducting state of elemen- tal scandium at a pressure of 260 gpa, Phys. Rev. Lett. 130, 256002 (2023)
2023
-
[23]
Matsuoka, T
T. Matsuoka, T. Ishikawa, and K. Shimizu, Superconduc- tivity of elements, Journal of Physics: Condensed Matter 38, 133001 (2026)
2026
-
[24]
N. C. Holmes, J. A. Moriarty, G. R. Gathers, and W. J. Nellis, The equation of state of platinum to 660 gpa (6.6 mbar), Journal of Applied Physics66, 2962 (1989)
1989
-
[25]
A. P. Hammersley, S. O. Svensson, M. Hanfland, A. N. Fitch, and D. Hausermann, Two-dimensional detector software: From real detector to idealised image or two- theta scan, High Pressure Research14, 235 (1996)
1996
-
[26]
Akahama and H
Y. Akahama and H. Kawamura, High-pressure raman spectroscopy of diamond anvils to 250gpa: Method for pressure determination in the multimegabar pressure range, Journal of Applied Physics96, 3748 (2004)
2004
-
[27]
E. V. Podryabinkin, E. V. Tikhonov, A. V. Shapeev, and A. R. Oganov, Accelerating crystal structure prediction by machine-learning interatomic potentials with active learning, Phys. Rev. B99, 064114 (2019)
2019
-
[28]
C. J. Pickard, Ephemeral data derived potentials for ran- dom structure search, Phys. Rev. B106, 014102 (2022)
2022
-
[29]
Ishikawa, Y
T. Ishikawa, Y. Tanaka, and S. Tsuneyuki, Evolution- ary search for superconducting phases in the lanthanum- nitrogen-hydrogen system with universal neural network potential, Phys. Rev. B109, 094106 (2024)
2024
-
[30]
Ishikawa, Y
T. Ishikawa, Y. Tanaka, and S. Tsuneyuki, Evolution- ary search for superconducting hydrides in the la-pt-h ternary system under compression, Physica Scripta101, 105903 (2026)
2026
-
[31]
H. Putz, J. C. Sch¨ on, and M. Jansen, Combined method forab initiostructure solution from powder diffraction data, Journal of Applied Crystallography32, 864 (1999)
1999
-
[32]
O. J. Lanning, S. Habershon, K. D. Harris, R. L. John- ston, B. M. Kariuki, E. Tedesco, and G. W. Turner, Defi- nition of a ‘guiding function’ in global optimization: a hy- brid approach combining energy and r-factor in structure solution from powder diffraction data, Chemical Physics Letters317, 296 (2000)
2000
-
[33]
A. A. Coelho, Whole-profile structure solution from pow- der diffraction data using simulated annealing, Journal of Applied Crystallography33, 899 (2000)
2000
-
[34]
Meredig and C
B. Meredig and C. Wolverton, A hybrid computational– experimental approach for automated crystal structure solution, Nature Materials12, 123 (2013)
2013
-
[35]
P. Gao, Q. Tong, J. Lv, Y. Wang, and Y. Ma, X-ray diffraction data-assisted structure searches, Computer Physics Communications213, 40 (2017)
2017
-
[36]
L. Ward, K. Michel, and C. Wolverton, Automated crys- tal structure solution from powder diffraction data: Val- idation of the first-principles-assisted structure solution method, Phys. Rev. Mater.1, 063802 (2017)
2017
-
[37]
Adachi, N
D. Adachi, N. Tsujimoto, R. Akashi, S. Todo, and S. Tsuneyuki, Search for common minima in joint op- timization of multiple cost functions, Computer Physics Communications241, 92 (2019)
2019
-
[38]
J. Lee, J. Oba, N. Ohba, and S. Kajita, Creation of crys- tal structure reproducing x-ray diffraction pattern with- out using database, npj Computational Materials9, 135 (2023)
2023
-
[39]
Tsujimoto, D
N. Tsujimoto, D. Adachi, R. Akashi, S. Todo, and S. Tsuneyuki, Crystal structure prediction supported by incomplete experimental data, Phys. Rev. Mater.2, 053801 (2018)
2018
-
[40]
Yoshikawa, R
S. Yoshikawa, R. Sato, R. Akashi, S. Todo, and S. Tsuneyuki, A noise-robust data assimilation method for crystal structure determination using powder diffrac- tion intensity, The Journal of Chemical Physics157, 224112 (2022)
2022
-
[41]
Y. Zhao, R. Sato, and S. Tsuneyuki, Accelerating sim- ulated annealing of glassy materials with data assim- ilation, Journal of Non-Crystalline Solids600, 122028 (2023)
2023
-
[42]
Zarrouk, R
T. Zarrouk, R. Ibragimova, A. P. Bart´ ok, and M. A. Caro, Experiment-driven atomistic materials modeling: 11 A case study combining x-ray photoelectron spectroscopy and machine learning potentials to infer the structure of oxygen-rich amorphous carbon, Journal of the American Chemical Society146, 14645 (2024)
2024
-
[43]
Y. Kubo, R. Sato, Y. Zhao, T. Ishikawa, and S. Tsuneyuki, Data-assimilated crystal growth simulation for multiple crystalline phases, The Journal of Chemical Physics161, 214113 (2024)
2024
-
[44]
Racioppi, A
S. Racioppi, A. Otero-de-la Roza, S. Hajinazar, and E. Zurek, Powder x-ray diffraction assisted evolutionary algorithm for crystal structure prediction, Digital Discov- ery4, 73 (2025)
2025
-
[45]
T. Zarrouk and M. A. Caro, Molecular augmented dy- namics: Generating experimentally consistent atomistic structures by design (2025), arXiv:2508.17132
-
[46]
Takamoto, C
S. Takamoto, C. Shinagawa, D. Motoki, K. Nak- ago, W. Li, I. Kurata, T. Watanabe, Y. Yayama, H. Iriguchi, Y. Asano, T. Onodera, T. Ishii, T. Kudo, H. Ono, R. Sawada, R. Ishitani, M. Ong, T. Yam- aguchi, T. Kataoka, A. Hayashi, N. Charoenphakdee, and T. Ibuka, Towards universal neural network potential for material discovery applicable to arbitrary combina...
2022
-
[47]
Matlantis, software as a service style material discovery tool,https://matlantis.com/
-
[48]
Thompson, L
A. Thompson, L. Swiler, C. Trott, S. Foiles, and G. Tucker, Spectral neighbor analysis method for auto- mated generation of quantum-accurate interatomic po- tentials, Journal of Computational Physics285, 316 (2015)
2015
-
[49]
M. A. Wood and A. P. Thompson, Extending the accu- racy of the snap interatomic potential form, The Journal of Chemical Physics148, 241721 (2018)
2018
-
[50]
Rohskopf, C
A. Rohskopf, C. Sievers, N. Lubbers, M. Cusentino, J. Goff, J. Janssen, M. McCarthy, D. M. O. de Za- piain, S. Nikolov, K. Sargsyan, D. Sema, E. Sikorski, L. Williams, A. Thompson, and M. Wood, Fitsnap: Atomistic machine learning with lammps, Journal of Open Source Software8, 5118 (2023)
2023
-
[51]
P. E. Bl¨ ochl, Projector augmented-wave method, Phys. Rev. B50, 17953 (1994)
1994
-
[52]
Kresse and D
G. Kresse and D. Joubert, From ultrasoft pseudopoten- tials to the projector augmented-wave method, Phys. Rev. B59, 1758 (1999)
1999
-
[53]
Kresse and J
G. Kresse and J. Furthm¨ uller, Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set, Computational Materials Science 6, 15 (1996)
1996
-
[54]
See Supplemental Material at xxx for additional compu- tational details, including the construction and validation of the qSNAP training set, fitting parameters, and rep- resentative data-assimilation-derived structures prior to DFT relaxation
-
[55]
Kirkpatrick, C
S. Kirkpatrick, C. D. Gelatt, and M. P. Vecchi, Optimiza- tion by simulated annealing, Science220, 671 (1983)
1983
-
[56]
Woodcock, Isothermal molecular dynamics calcula- tions for liquid salts, Chemical Physics Letters10, 257 (1971)
L. Woodcock, Isothermal molecular dynamics calcula- tions for liquid salts, Chemical Physics Letters10, 257 (1971)
1971
-
[57]
Plimpton, Fast parallel algorithms for short-range molecular dynamics, Journal of Computational Physics 117, 1 (1995)
S. Plimpton, Fast parallel algorithms for short-range molecular dynamics, Journal of Computational Physics 117, 1 (1995)
1995
-
[58]
A. P. Thompson, H. M. Aktulga, R. Berger, D. S. Bolin- tineanu, W. M. Brown, P. S. Crozier, P. J. in ’t Veld, A. Kohlmeyer, S. G. Moore, T. D. Nguyen, R. Shan, M. J. Stevens, J. Tranchida, C. Trott, and S. J. Plimpton, Lammps - a flexible simulation tool for particle-based materials modeling at the atomic, meso, and continuum scales, Computer Physics Comm...
2022
-
[59]
Giannozzi, S
P. Giannozzi, S. Baroni, N. Bonini, M. Calandra, R. Car, C. Cavazzoni, D. Ceresoli, G. L. Chiarotti, M. Cococ- cioni, I. Dabo, A. Dal Corso, S. de Gironcoli, S. Fabris, G. Fratesi, R. Gebauer, U. Gerstmann, C. Gougoussis, A. Kokalj, M. Lazzeri, L. Martin-Samos, N. Marzari, F. Mauri, R. Mazzarello, S. Paolini, A. Pasquarello, L. Paulatto, C. Sbraccia, S. S...
2009
-
[60]
Vanderbilt, Soft self-consistent pseudopotentials in a generalized eigenvalue formalism, Phys
D. Vanderbilt, Soft self-consistent pseudopotentials in a generalized eigenvalue formalism, Phys. Rev. B41, 7892 (1990)
1990
-
[61]
H. T. Stokes and D. M. Hatch,FINDSYM: program for identifying the space-group symmetry of a crystal, Jour- nal of Applied Crystallography38, 237 (2005)
2005
-
[62]
W. L. McMillan, Transition temperature of strong- coupled superconductors, Phys. Rev.167, 331 (1968)
1968
-
[63]
Dynes, Mcmillan’s equation and the tc of supercon- ductors, Solid State Communications10, 615 (1972)
R. Dynes, Mcmillan’s equation and the tc of supercon- ductors, Solid State Communications10, 615 (1972)
1972
-
[64]
P. B. Allen and R. C. Dynes, Transition temperature of strong-coupled superconductors reanalyzed, Phys. Rev. B12, 905 (1975)
1975
-
[65]
B. H. Toby and R. B. Von Dreele,GSAS-II: the gene- sis of a modern open-source all purpose crystallography software package, Journal of Applied Crystallography46, 544 (2013)
2013
-
[66]
Momma and F
K. Momma and F. Izumi,VESTA3for three-dimensional visualization of crystal, volumetric and morphology data, Journal of Applied Crystallography44, 1272 (2011)
2011
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.