PhAME: Phenotype-Aware Molecular Editing via Latent Diffusion
Pith reviewed 2026-06-29 13:34 UTC · model grok-4.3
The pith
PhAME edits molecules in a VAE latent space using two independent classifier-free guidance scales to balance phenotypic targets against structural similarity to a seed.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
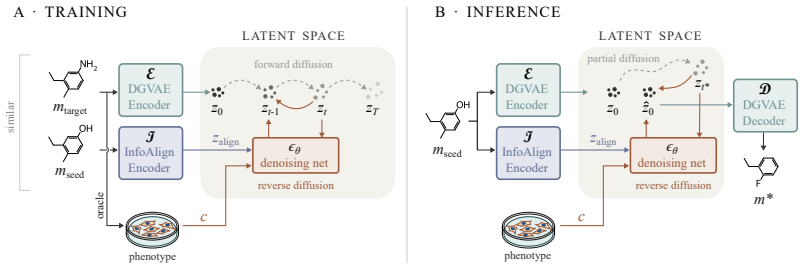
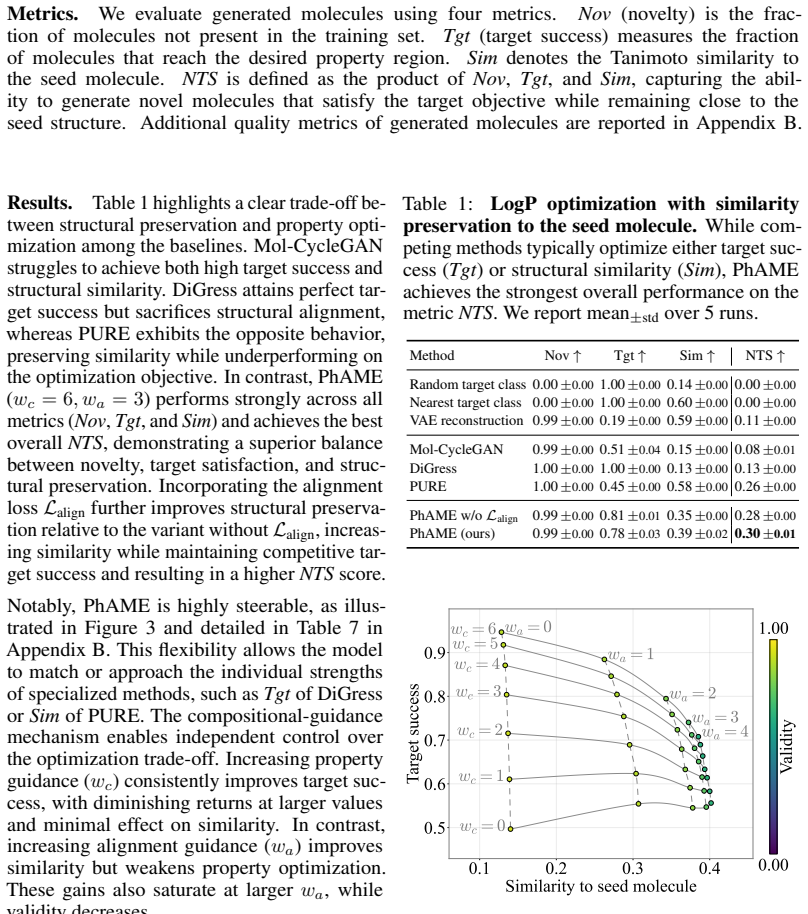
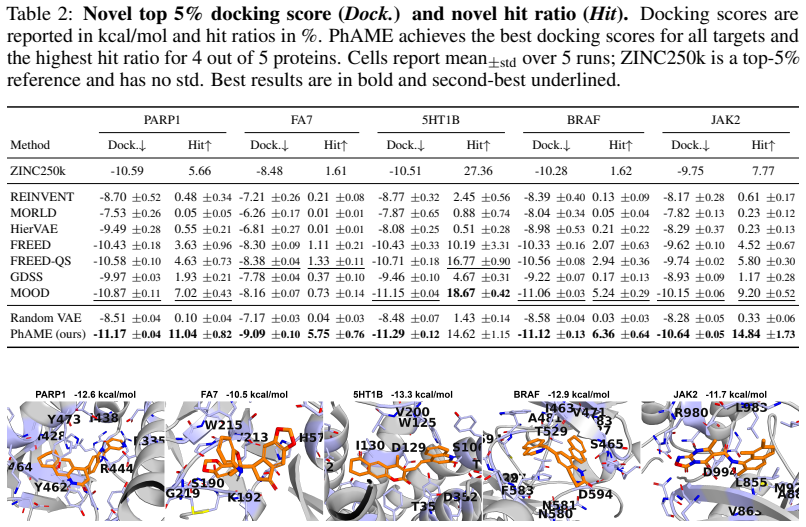
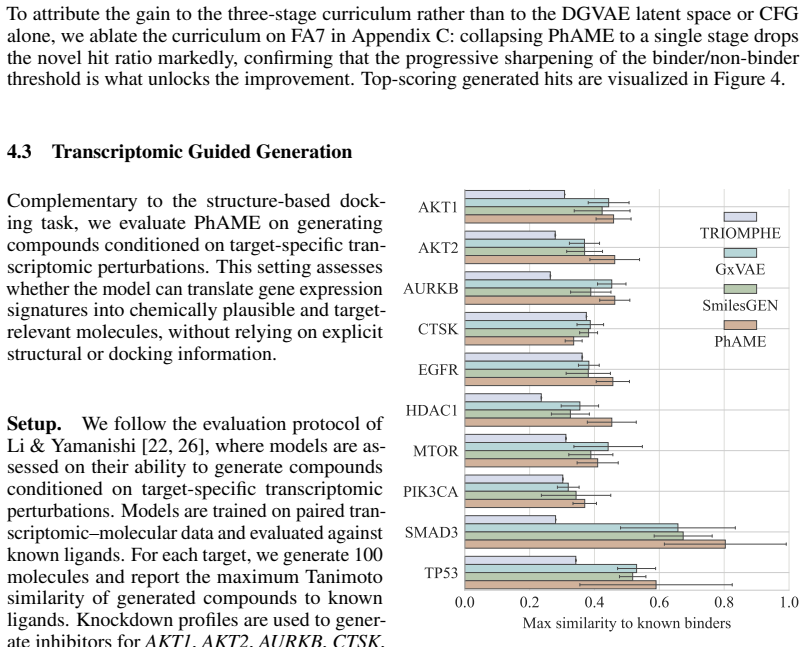
PhAME recasts molecular optimization as editing in the latent space of a pretrained graph-based VAE. Its central contribution is a compositional classifier-free guidance scheme with two independent scales, one for the phenotype-conditioning and one for similarity to the seed structure, allowing practitioners to control the tradeoff between these two objectives. Empirical evaluations across diverse benchmarks, including docking score optimization and multimodal phenotypic generation, demonstrate that PhAME achieves state-of-the-art results while maintaining high chemical validity and novelty.
What carries the argument
Compositional classifier-free guidance scheme with two independent scales, one controlling phenotype-conditioning strength and the other controlling similarity to the seed structure.
If this is right
- Practitioners gain explicit control over the tradeoff between achieving desired phenotypic signatures and maintaining proximity to a known hit.
- The method produces state-of-the-art performance on docking score optimization tasks.
- It supports multimodal phenotypic generation while keeping high chemical validity and novelty.
- Molecular editing becomes possible without sacrificing either biological relevance or structural closeness to the starting molecule.
Where Pith is reading between the lines
- Similar dual-scale guidance could be tested on other latent generative models beyond graph VAEs, such as those operating on SMILES or 3D conformers.
- The approach may reduce the number of synthesis rounds needed in hit-to-lead campaigns by allowing finer control over how far an edit moves from the original molecule.
- If the two scales remain independent across different phenotype data types, the framework could support joint optimization of cell morphology and transcriptomic readouts without additional retraining.
Load-bearing premise
The latent space of the pretrained graph-based VAE supports meaningful, controllable edits that simultaneously respect phenotypic signatures and structural proximity.
What would settle it
Generation runs in which increasing the phenotype guidance scale fails to improve phenotypic match while the structure scale fails to preserve seed similarity, or where the two scales cannot be varied independently without one dominating the other.
Figures

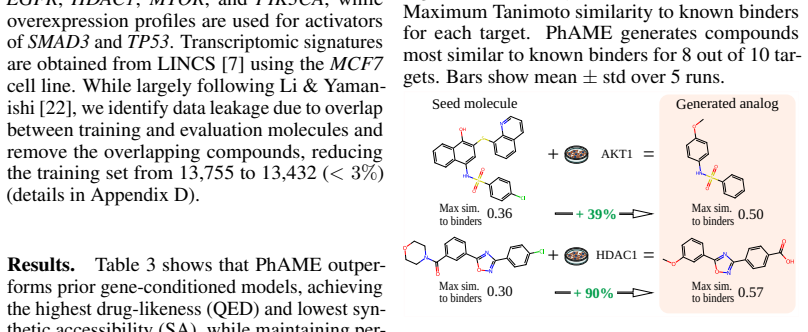
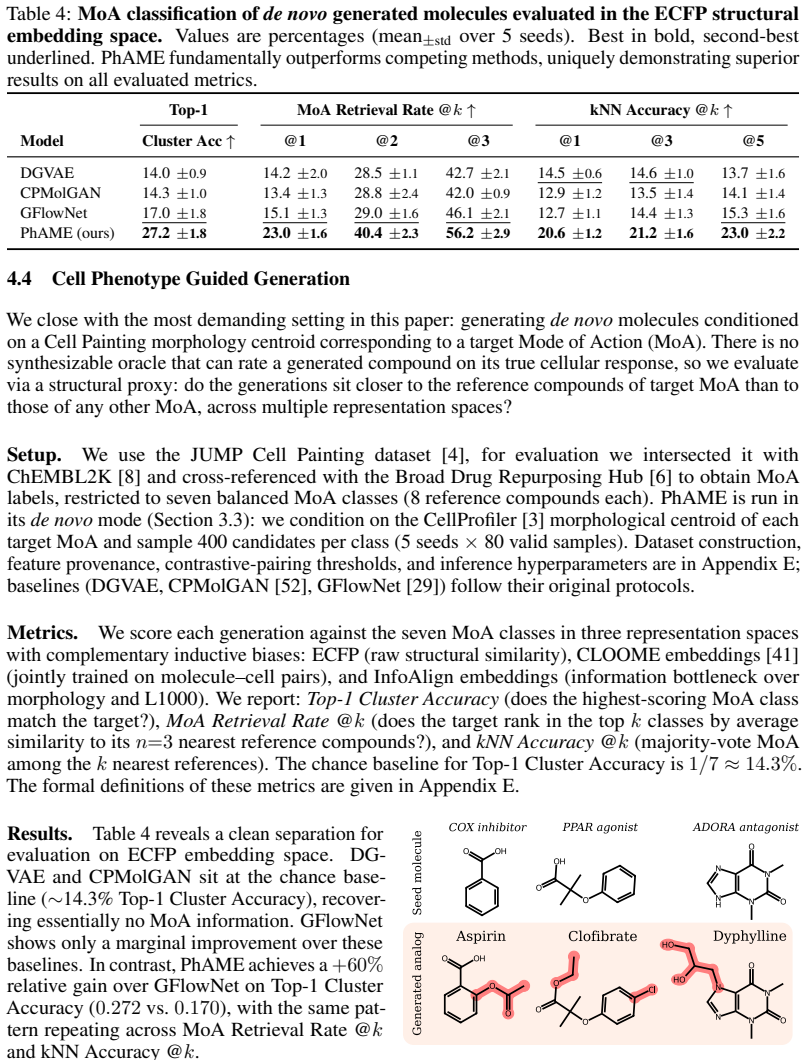

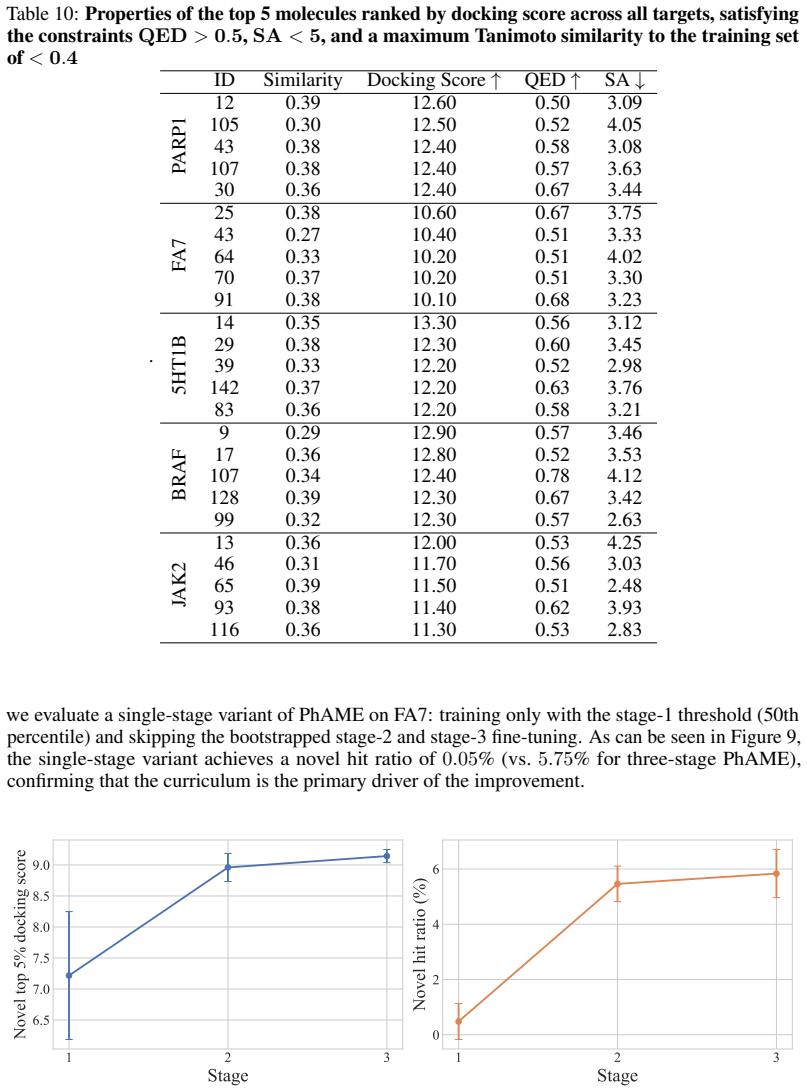

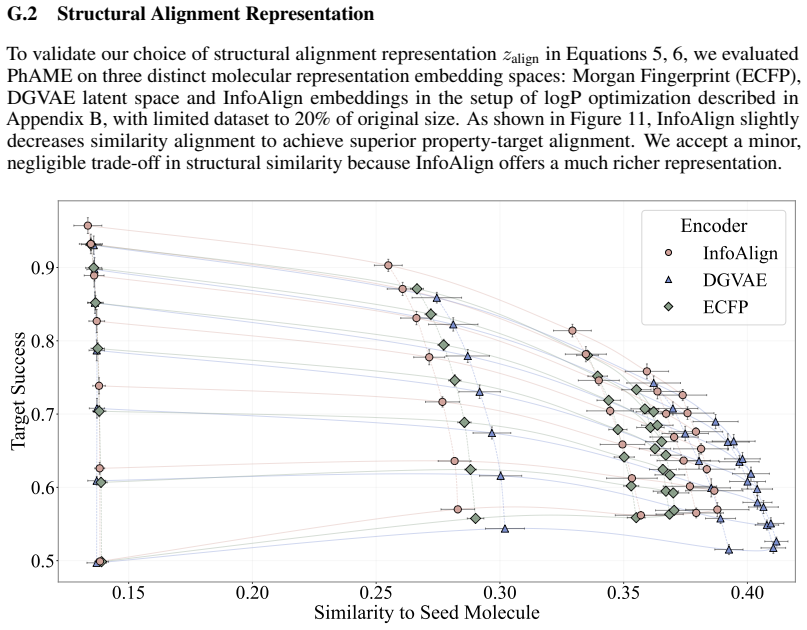

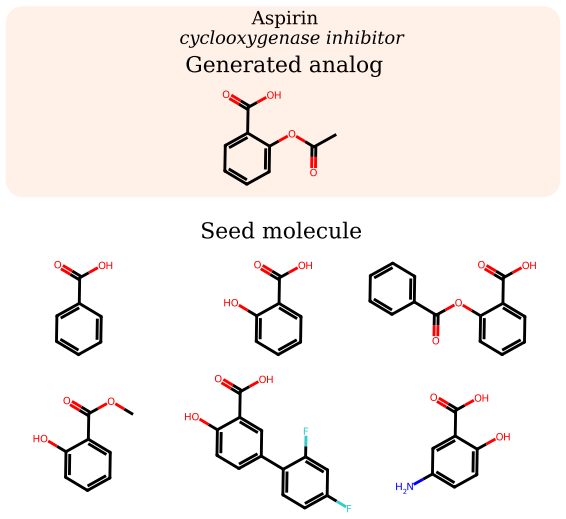
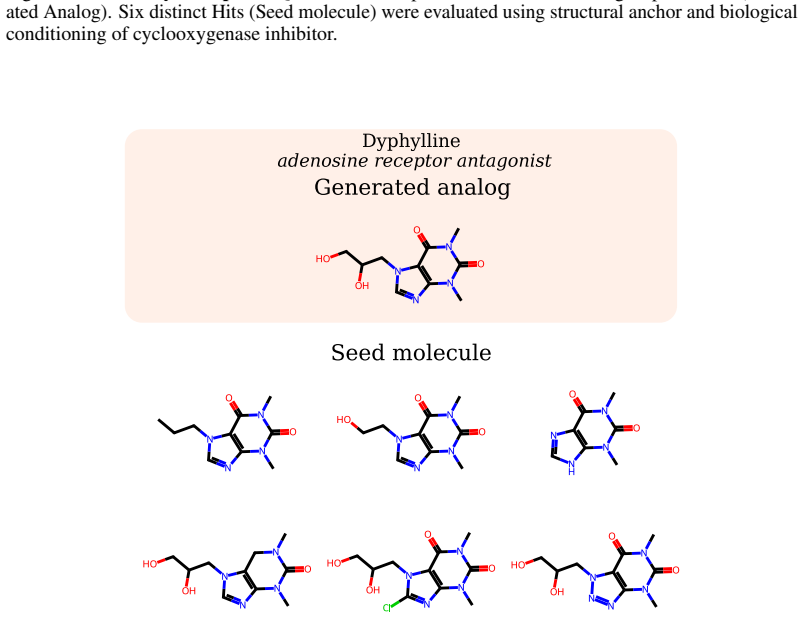
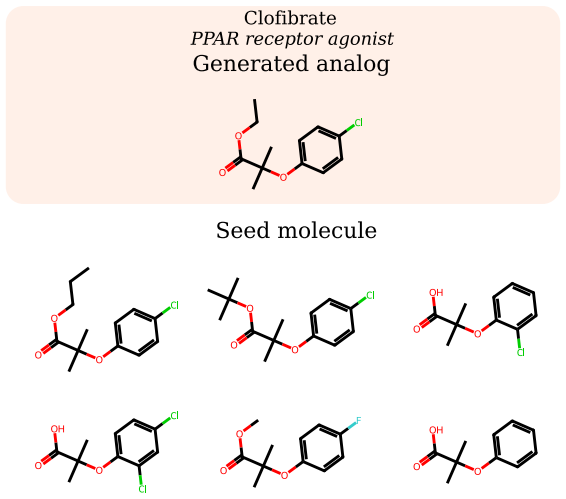
read the original abstract
Small-molecule drug discovery requires simultaneous optimization of numerous properties of candidate molecules. These properties can be investigated through the analysis of high-dimensional biological signatures, such as cell morphology and transcriptomic perturbations, which provide a rich perspective on the underlying biological mechanisms. However, existing generative methods, which use those signatures for optimization, fail to meet two key requirements: providing precise guidance toward desired phenotypic signatures while maintaining structural proximity to a known hit. We introduce PhAME (Phenotype-Aware Molecular Editing), a latent diffusion framework that overcomes this challenge by recasting molecular optimization as editing in the latent space of a pretrained graph-based VAE. Our central contribution is a compositional classifier-free guidance scheme with two independent scales, one for the phenotype-conditioning and one for similarity to the seed structure, allowing practitioners to control the tradeoff between these two objectives. Empirical evaluations across diverse benchmarks, including docking score optimization and multimodal phenotypic generation, demonstrate that PhAME achieves state-of-the-art results while maintaining high chemical validity and novelty.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The paper introduces PhAME, a latent diffusion framework for molecular editing that recasts optimization as editing in the latent space of a pretrained graph-based VAE. Its central contribution is a compositional classifier-free guidance scheme using two independent scales—one for phenotype conditioning and one for similarity to the seed structure—to control the tradeoff between phenotypic signatures and structural proximity. The work claims state-of-the-art empirical results on docking score optimization and multimodal phenotypic generation benchmarks while preserving high chemical validity and novelty.
Significance. If the two-scale guidance scheme successfully enables independent control without feature entanglement, the method would offer a practical advance for multi-objective small-molecule optimization in drug discovery by allowing tunable tradeoffs between biological phenotype matching and retention of known hit structures. The approach leverages standard classifier-free guidance in a compositional manner on top of an existing VAE, which is a reasonable extension, but its impact hinges on whether the frozen VAE latent space actually supports the required separation.
major comments (1)
- [Abstract] Abstract: The central claim that the compositional classifier-free guidance with two independent scales allows controllable tradeoffs presupposes that the pretrained graph VAE latent space contains factors that can be modulated separately for phenotypic signatures versus structural proximity to the seed. Standard graph VAEs are trained solely on molecular graphs without phenotype supervision, so phenotype information is likely entangled with structural features; the manuscript provides no explicit mechanism, loss term, or post-hoc analysis to guarantee orthogonality or independent controllability of the two conditioning signals once the diffusion model is trained on the frozen VAE. This assumption is load-bearing for the claimed advantage over prior generative methods.
Simulated Author's Rebuttal
We thank the referee for the careful reading and for identifying the key assumption underlying our central contribution. We respond to the major comment below.
read point-by-point responses
-
Referee: [Abstract] Abstract: The central claim that the compositional classifier-free guidance with two independent scales allows controllable tradeoffs presupposes that the pretrained graph VAE latent space contains factors that can be modulated separately for phenotypic signatures versus structural proximity to the seed. Standard graph VAEs are trained solely on molecular graphs without phenotype supervision, so phenotype information is likely entangled with structural features; the manuscript provides no explicit mechanism, loss term, or post-hoc analysis to guarantee orthogonality or independent controllability of the two conditioning signals once the diffusion model is trained on the frozen VAE. This assumption is load-bearing for the claimed advantage over prior generative methods.
Authors: We agree that the pretrained graph VAE is unsupervised with respect to phenotype and that the manuscript does not supply an explicit loss term or theoretical guarantee of orthogonality between phenotypic and structural factors in the latent space. The method instead demonstrates practical controllability through the compositional classifier-free guidance applied at sampling time: the diffusion model is trained to predict noise under both phenotype and seed-structure conditions, after which independent guidance scales are used to steer the two objectives. Our empirical results across docking-score optimization and multimodal phenotypic benchmarks show that varying the two scales produces the expected tradeoffs in phenotypic fidelity versus structural similarity while preserving validity. We acknowledge the absence of post-hoc analysis of latent-space entanglement and will add, in revision, both a discussion of this assumption and new experiments that quantify correlations between phenotype predictions and structural features in the VAE latent space together with further guidance-scale ablations. revision: partial
Circularity Check
No significant circularity; derivation is self-contained methodological contribution
full rationale
The paper introduces a latent diffusion model with a new compositional classifier-free guidance scheme using two independent scales. No equations, fitting procedures, or self-citations are visible that reduce the central claim to an input by construction. The method is framed as a novel recasting of optimization as editing in a pretrained VAE latent space, with empirical evaluations presented as external validation. This matches the default expectation of no circularity.
Axiom & Free-Parameter Ledger
Reference graph
Works this paper leans on
-
[1]
Alhossary, S
A. Alhossary, S. D. Handoko, Y . Mu, and C.-K. Kwoh. Fast, accurate, and reliable molecular docking with quickvina 2.Bioinformatics, 31(13):2214–2216, 2015
2015
-
[2]
M.-A. Bray, S. Singh, H. Han, C. T. Davis, B. Borgeson, C. Hartland, M. Kost-Alimova, S. M. Gustafsdottir, C. C. Gibson, and A. E. Carpenter. Cell painting, a high-content image- based assay for morphological profiling using multiplexed fluorescent dyes.Nature protocols, 11(9):1757–1774, 2016
2016
-
[3]
A. E. Carpenter, T. R. Jones, M. R. Lamprecht, C. Clarke, I. H. Kang, O. Friman, D. A. Guertin, J. H. Chang, R. A. Lindquist, J. Moffat, P. Golland, and D. M. Sabatini. Cellprofiler: image analysis software for identifying and quantifying cell phenotypes. 2006
2006
-
[4]
S. N. Chandrasekaran, J. Ackerman, E. Alix, D. M. Ando, J. Arevalo, M. Bennion, N. Boisseau, A. Borowa, J. D. Boyd, and e. a. Brino. Jump cell painting dataset: morphological impact of 136,000 chemical and genetic perturbations.bioRxiv, 2023
2023
-
[5]
S. N. Chandrasekaran, H. Ceulemans, J. D. Boyd, and A. E. Carpenter. Image-based profiling for drug discovery: due for a machine-learning upgrade?Nature reviews drug discovery, 20(2):145–159, 2021
2021
-
[6]
S. M. Corsello, J. A. Bittker, Z. Liu, J. Gould, P. McCarren, J. E. Hirschman, S. E. Johnston, A. Vrcic, B. Wong, M. Khan, J. Asiedu, R. Narayan, C. C. Mader, A. Subramanian, and T. R. Golub. The drug repurposing hub: a next-generation drug library and information resource. Nature medicine, 2017. 10
2017
-
[7]
Q. Duan, C. Flynn, M. Niepel, M. Hafner, J. L. Muhlich, N. F. Fernandez, A. D. Rouillard, C. M. Tan, E. Y . Chen, T. R. Golub, et al. Lincs canvas browser: interactive web app to query, browse and interrogate lincs l1000 gene expression signatures.Nucleic acids research, 42(W1):W449–W460, 2014
2014
-
[8]
Gaulton, L
A. Gaulton, L. J. Bellis, A. P. Bento, J. Chambers, M. Davies, A. Hersey, Y . Light, S. McGlinchey, D. Michalovich, B. Al-Lazikani, and J. P. Overington. Chembl: a large-scale bioactivity database for drug discovery.Nucleic Acids Research, 2012
2012
-
[9]
Gómez-Bombarelli, J
R. Gómez-Bombarelli, J. N. Wei, D. Duvenaud, J. M. Hernández-Lobato, B. Sánchez-Lengeling, D. Sheberla, J. Aguilera-Iparraguirre, T. D. Hirzel, R. P. Adams, and A. Aspuru-Guzik. Auto- matic chemical design using a data-driven continuous representation of molecules.ACS central science, 4(2):268–276, 2018
2018
-
[10]
Gupta, B
A. Gupta, B. Lenin, S. Current, R. Batra, B. Ravindran, K. Raman, and S. Parthasarathy. Pure: policy-guided unbiased representations for structure-constrained molecular generation.Journal of Cheminformatics, 17(1):1–18, 2025
2025
-
[11]
J. Ho, A. Jain, and P. Abbeel. Denoising diffusion probabilistic models. In H. Larochelle, M. Ranzato, R. Hadsell, M. Balcan, and H. Lin, editors,Advances in Neural Information Processing Systems, volume 33, pages 6840–6851. Curran Associates, Inc., 2020
2020
-
[12]
Ho and T
J. Ho and T. Salimans. Classifier-free diffusion guidance, 2022
2022
-
[13]
Hoogeboom, V
E. Hoogeboom, V . G. Satorras, C. Vignac, and M. Welling. Equivariant diffusion for molecule generation in 3d. InInternational conference on machine learning, pages 8867–8887. PMLR, 2022
2022
-
[14]
J. J. Irwin and B. K. Shoichet. Zinc- a free database of commercially available compounds for virtual screening.Journal of chemical information and modeling, 45(1):177–182, 2005
2005
-
[15]
Jiang, X
Q. Jiang, X. Ye, Z. Guo, Y . Xia, Z. Liu, J. Xu, P. Jin, F. Ju, H. Xia, S. Feng, et al. Phenotype- guided in silico molecular generation using large language models.bioRxiv, pages 2026–01, 2026
2026
-
[16]
W. Jin, R. Barzilay, and T. Jaakkola. Junction tree variational autoencoder for molecular graph generation. InInternational conference on machine learning, pages 2323–2332. PMLR, 2018
2018
-
[17]
B. Jing, G. Corso, J. Chang, R. Barzilay, and T. Jaakkola. Torsional diffusion for molecular conformer generation.Advances in neural information processing systems, 35:24240–24253, 2022
2022
-
[18]
Kaitoh and Y
K. Kaitoh and Y . Yamanishi. Triomphe: Transcriptome-based inference and generation of molecules with desired phenotypes by machine learning.Journal of Chemical Information and Modeling, 61(9):4303–4320, 2021
2021
-
[19]
S. Lee, J. Jo, and S. J. Hwang. Exploring chemical space with score-based out-of-distribution generation. InInternational Conference on Machine Learning, pages 18872–18892. PMLR, 2023
2023
- [20]
-
[21]
Li and Y
C. Li and Y . Yamanishi. Gxvaes: Two joint vaes generate hit molecules from gene expression profiles. InProceedings of the AAAI Conference on Artificial Intelligence, volume 38, pages 13455–13463, 2024
2024
-
[22]
Li and Y
C. Li and Y . Yamanishi. Gxvaes: Two joint vaes generate hit molecules from gene expression profiles.Proceedings of the AAAI Conference on Artificial Intelligence, 38(12):13455–13463, Mar. 2024
2024
-
[23]
Z. Liang, S. Zhuang, X. Jiao, W. Mao, H. Chen, and C. Shen. scppdm: A diffusion model for single-cell drug-response prediction.arXiv preprint arXiv:2510.11726, 2025. 11
-
[24]
G. Liu, S. Seal, J. Arevalo, Z. Liang, A. E. Carpenter, M. Jiang, and S. Singh. Learning molecular representation in a cell. InThe Thirteenth International Conference on Learning Representations, 2025
2025
- [25]
-
[26]
H. Liu, S. Tian, and X. Liu. Phenotypic profile-informed generation of drug-like molecules via dual-channel variational autoencoders. InProceedings of the Thirty-Fourth International Joint Conference on Artificial Intelligence, IJCAI ’25, 2025
2025
- [27]
-
[28]
H. H. Loeffler, J. He, A. Tibo, J. P. Janet, A. V oronov, L. H. Mervin, and O. Engkvist. Reinvent 4: Modern ai–driven generative molecule design.Journal of Cheminformatics, 16(1):20, 2024
2024
-
[29]
S. Z. Lu, Z. Lu, E. Hajiramezanali, T. Biancalani, Y . Bengio, G. Scalia, and M. Koziarski. Cell morphology-guided small molecule generation with gflownets. InICML 2024 Workshop on Structured Probabilistic Inference{\&}Generative Modeling, 2024
2024
-
[30]
Maziarka, A
Ł. Maziarka, A. Pocha, J. Kaczmarczyk, K. Rataj, T. Danel, and M. Warchoł. Mol-cyclegan: a generative model for molecular optimization.Journal of Cheminformatics, 12(1):2, 2020
2020
-
[31]
Méndez-Lucio, B
O. Méndez-Lucio, B. Baillif, D.-A. Clevert, D. Rouquié, and J. Wichard. De novo generation of hit-like molecules from gene expression signatures using artificial intelligence.Nature communications, 11(1):10, 2020
2020
-
[32]
F. M. Morelli, V . Kim, F. Hecker, S. Geibel, and P. A. Marín Zapata. unidino: Assay-independent feature extraction for fluorescence microscopy images.Computational and Structural Biotech- nology Journal, 27:928–936, 2025
2025
-
[33]
Moshkov, M
N. Moshkov, M. Bornholdt, S. Benoit, M. Smith, C. McQuin, A. Goodman, R. A. Senft, Y . Han, M. Babadi, P. Horvath, B. A. Cimini, A. E. Carpenter, S. Singh, and J. C. Caicedo. Learning representations for image-based profiling of perturbations.bioRxiv, 2022
2022
-
[34]
Nguyen and A
T. Nguyen and A. Karolak. Expanding molecular design with graph variational autoencoders: A comparative study of pair-encoding and character tokenization.ChemRxiv, 2025(0523), 2025
2025
-
[35]
S. K. Niazi. The coming of age of ai/ml in drug discovery, development, clinical testing, and manufacturing: the fda perspectives.Drug Design, Development and Therapy, pages 2691–2725, 2023
2023
-
[36]
A. Q. Nichol and P. Dhariwal. Improved denoising diffusion probabilistic models. InInterna- tional conference on machine learning, pages 8162–8171. PMLR, 2021
2021
-
[37]
International Conference on Learning Representations , year=
A. Palma, T. Richter, H. Zhang, M. Lubetzki, A. Tong, A. Dittadi, and F. Theis. Multi-modal and multi-attribute generation of single cells with cfgen.arXiv preprint arXiv:2407.11734, 2024
-
[38]
P. G. Polishchuk, T. I. Madzhidov, and A. Varnek. Estimation of the size of drug-like chemical space based on gdb-17 data.Journal of computer-aided molecular design, 27(8):675–679, 2013
2013
-
[39]
Polykovskiy, A
D. Polykovskiy, A. Zhebrak, B. Sanchez-Lengeling, S. Golovanov, O. Tatanov, S. Belyaev, R. Kurbanov, A. Artamonov, V . Aladinskiy, M. Veselov, et al. Molecular sets (moses): a bench- marking platform for molecular generation models.Frontiers in pharmacology, 11:565644, 2020
2020
-
[40]
Rombach, A
R. Rombach, A. Blattmann, D. Lorenz, P. Esser, and B. Ommer. High-resolution image synthesis with latent diffusion models. InProceedings of the IEEE/CVF conference on computer vision and pattern recognition, pages 10684–10695, 2022
2022
-
[41]
Sanchez-Fernandez, E
A. Sanchez-Fernandez, E. Rumetshofer, S. Hochreiter, and G. Klambauer. Cloome: contrastive learning unlocks bioimaging databases for queries with chemical structures. 2023. 12
2023
-
[42]
J. Song, C. Meng, and S. Ermon. Denoising diffusion implicit models. InInternational Conference on Learning Representations, 2021
2021
-
[43]
R. Song and H. Liu. Phenomoler: Phenotype-guided molecular optimization via chemistry large language model.arXiv preprint arXiv:2509.21424, 2025
-
[44]
Y . Song, J. Sohl-Dickstein, D. P. Kingma, A. Kumar, S. Ermon, and B. Poole. Score-based generative modeling through stochastic differential equations. InInternational Conference on Learning Representations, 2021
2021
-
[45]
Subramanian, R
A. Subramanian, R. Narayan, S. M. Corsello, D. D. Peck, T. E. Natoli, X. Lu, J. Gould, J. F. Davis, A. A. Tubelli, J. K. Asiedu, D. L. Lahr, J. E. Hirschman, Z. Liu, M. Donahue, B. Julian, M. Khan, D. Wadden, I. C. Smith, D. Lam, A. Liberzon, C. Toder, M. Bagul, M. Orzechowski, O. M. Enache, F. Piccioni, S. A. Johnson, N. J. Lyons, A. H. Berger, A. F. Sha...
2017
-
[46]
D. C. Swinney. Phenotypic vs. target-based drug discovery for first-in-class medicines.Clinical Pharmacology & Therapeutics, 93(4):299–301, 2013
2013
-
[47]
Q. Tang, D. Ding, X. Yuan, G. Seabra, P. A. Ramdhan, C.-Y . Liu, M. T. Thai, C. Li, H. Luesch, and Y . Li. Mgmg: Cell morphology-guided molecule generation for drug discovery.bioRxiv, 2025
2025
-
[48]
Vignac, I
C. Vignac, I. Krawczuk, A. Siraudin, B. Wang, V . Cevher, and P. Frossard. Digress: Discrete denoising diffusion for graph generation. InProceedings of the 11th International Conference on Learning Representations, 2023
2023
-
[49]
C. Wang, H. H. Ong, S. Chiba, and J. C. Rajapakse. Gldm: hit molecule generation with constrained graph latent diffusion model.Briefings in bioinformatics, 25(3):bbae142, 2024
2024
-
[50]
Weiss, E
T. Weiss, E. Mayo Yanes, S. Chakraborty, L. Cosmo, A. M. Bronstein, and R. Gershoni-Poranne. Guided diffusion for inverse molecular design.Nature Computational Science, 3(10):873–882, 2023
2023
-
[51]
S. Yang, D. Hwang, S. Lee, S. Ryu, and S. J. Hwang. Hit and lead discovery with explorative rl and fragment-based molecule generation.Advances in Neural Information Processing Systems, 34:7924–7936, 2021
2021
-
[52]
P. A. M. Zapata, O. Méndez-Lucio, T. Le, C. J. Beese, J. Wichard, D. Rouquié, and D.-A. Clevert. Cell morphology-guided de novo hit design by conditioning gans on phenotypic image features.Digital discovery, 2(1):91–102, 2023
2023
-
[53]
C. Zeng, J. Jin, C. Ambrose, G. Karypis, M. Transtrum, E. B. Tadmor, R. G. Hennig, A. Roitberg, S. Martiniani, and M. Liu. Propmolflow: property-guided molecule generation with geometry- complete flow matching.Nature Computational Science, pages 1–10, 2026
2026
-
[54]
Zhang, J
Q. Zhang, J. Xiao, D. Niu, Z. Zhang, S. Ding, and Z. Li. Geometry-complete latent diffusion model for 3d molecule generation.Bioinformatics, 41(8):btaf426, 2025. 13 A Training Details Python environment.All experiments were carried out using Python 3.11.5. The key libraries are listed in Table 5. Other requirements are included in the code repository. Tab...
2025
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.