Excited States from Restricted Open Shell Plane-Wave DFT
Pith reviewed 2026-06-29 09:26 UTC · model grok-4.3
The pith
Plane-wave restricted open-shell Kohn-Sham DFT computes spin-pure singlet excitations and forces with accuracy comparable to TDDFT.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
The central claim is that the ROKS functional, implemented via preconditioned conjugate-gradient or DIIS optimization in the plane-wave PAW setting, yields excitation energies and excited-state forces whose accuracy is similar to that of TDDFT while preserving the scaling of ordinary ground-state DFT, as demonstrated by close numerical agreement on both molecular test cases and the MgO oxygen-vacancy defect.
What carries the argument
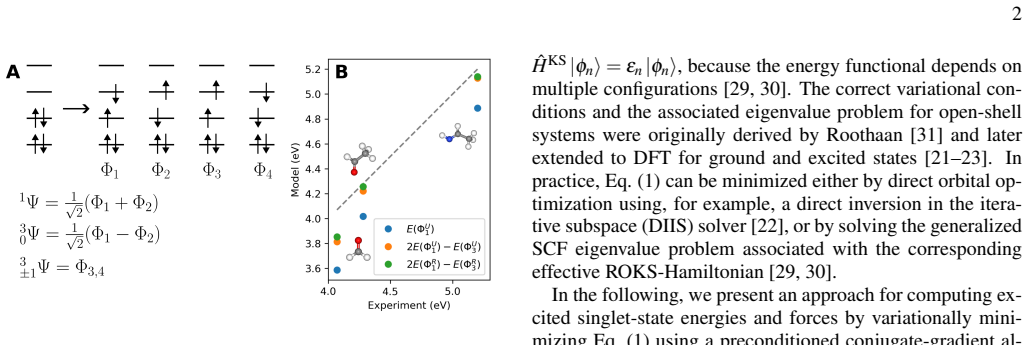
The ROKS energy functional, formed as a weighted average of mixed-spin and triplet configurations and minimized in the plane-wave PAW representation, together with the derived analytical atomic forces.
If this is right
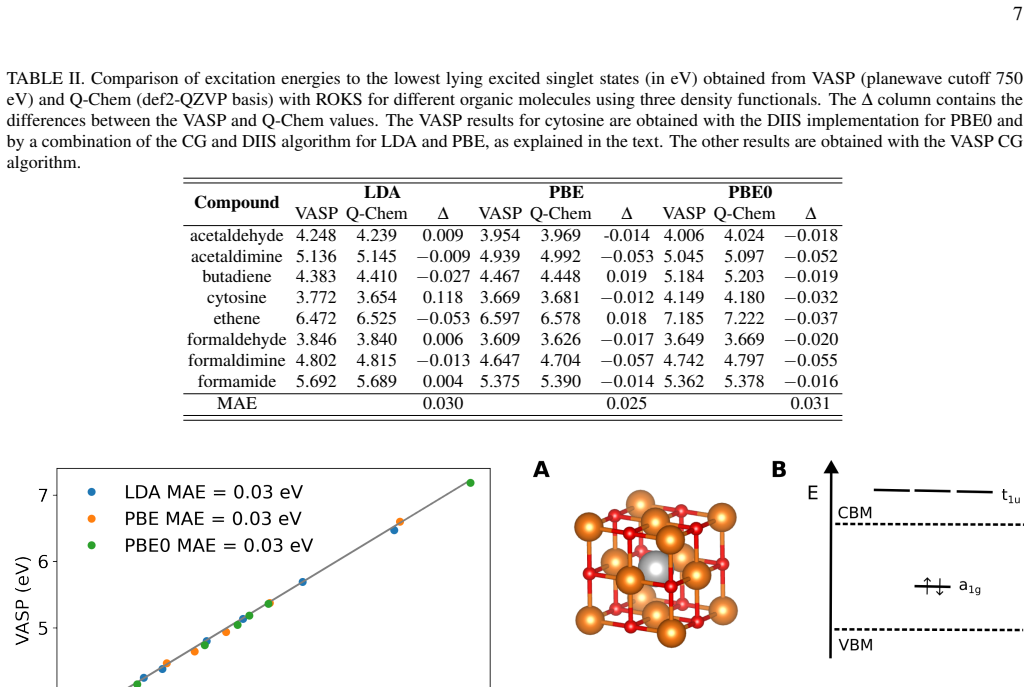
- ROKS excitation energies for the eight organic molecules agree with the Q-Chem reference to a mean deviation of approximately 30 meV.
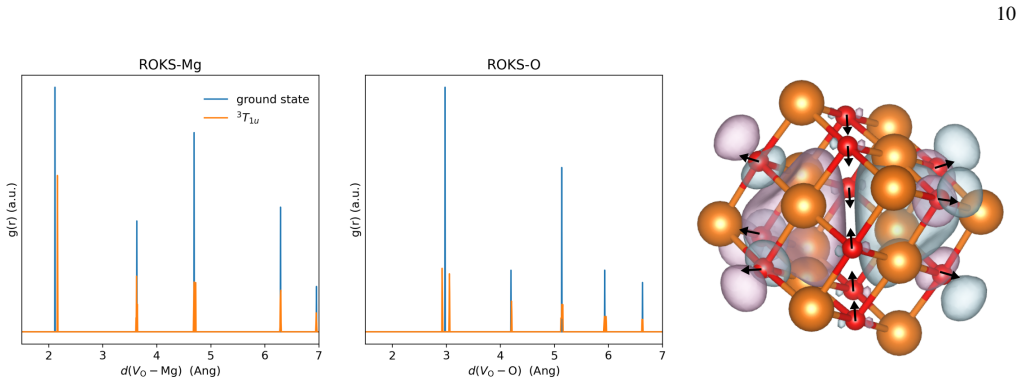
- For the MgO oxygen vacancy, vertical excitation energies from ROKS and TDDFT differ by 0.21 eV on average with a dielectric-dependent hybrid functional.
- Franck-Condon shifts and mass-weighted displacements between excited and ground states differ by 0.14 eV and 0.12 amu^{1/2} Ang on average between the two methods.
- Excited-state properties exhibit weaker dependence on the underlying DFT functional for ROKS than for TDDFT when the PBE functional is used.
- ROKS supplies both energies and forces at the computational cost of ground-state DFT, enabling simulations of excited states in extended systems.
Where Pith is reading between the lines
- The availability of analytical forces would permit routine excited-state geometry optimization and molecular dynamics in periodic defect systems where TDDFT scaling becomes prohibitive.
- Weaker functional dependence could reduce the need for expensive hybrid functionals in large supercell calculations of excited states.
- The method could be tested on additional point defects, surfaces, or low-dimensional materials to map its range of applicability beyond the MgO vacancy example.
- Direct comparison of ROKS results against higher-level wavefunction methods on the same periodic systems would quantify remaining systematic errors.
Load-bearing premise
The plane-wave PAW implementation of the ROKS functional converges to the same spin-pure singlet energies obtained by the Gaussian-basis ROKS code, without substantial extra errors from the basis representation or pseudopotential choice.
What would settle it
A side-by-side calculation of the eight organic-molecule excitation energies with both the new plane-wave code and the established Gaussian-basis ROKS implementation would show whether mean deviations remain near 30 meV or grow systematically.
Figures
read the original abstract
Variational excited-state density functional theory (DFT) enables the calculation of excited states at a cost comparable to ground-state calculations, but single-configuration approaches often suffer from spin contamination. We implement restricted open-shell Kohn-Sham (ROKS) DFT, which recovers spin-pure singlet excitation energies via the variational minimization of a weighted combination of mixed-spin and triplet configurations, within the plane-wave projector augmented-wave framework of VASP. The energy functional is optimized using a preconditioned conjugate-gradient or a direct inversion in the iterative subspace algorithm, and analytical atomic forces are derived. The implementation is validated for eight organic molecules by comparison to the Q-Chem quantum chemistry code, yielding mean deviations of approximately $30\,\mathrm{meV}$. As a solid-state application, we investigate the three lowest lying excitations of MgO with a neutral oxygen vacancy. For a dielectric-dependent hybrid functional, vertical excitation energies from ROKS and time-dependent density functional theory (TDDFT) differ on average by about $0.21\,\mathrm{eV}$. The Franck-Condon shifts deviate on average by $0.14\,\mathrm{eV}$ between the two methods and mass-weighted displacements between the excited states and the ground state by $0.12\,\mathrm{amu}^{1/2}$ Ang. Additional calculations at the PBE level reveal that these properties depend less strongly on the DFT functional for ROKS than for TDDFT. These results demonstrate that ROKS provides excitation energies and excited-state forces with an accuracy similar to TDDFT while retaining the favorable scaling of ground-state DFT, making it a promising approach for affordable excited-state simulations in extended systems.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript implements restricted open-shell Kohn-Sham (ROKS) DFT in the plane-wave PAW framework of VASP for variational excited-state calculations. It optimizes the ROKS energy functional with PCG or DIIS, derives analytical atomic forces, validates the implementation on eight organic molecules against Q-Chem (mean deviation ~30 meV), and applies it to the three lowest excitations of MgO with a neutral oxygen vacancy. Vertical excitation energies, Franck-Condon shifts, and mass-weighted displacements are compared to TDDFT (average differences 0.21 eV, 0.14 eV, 0.12 amu^{1/2} Å) using a dielectric-dependent hybrid functional; additional PBE calculations show weaker functional dependence for ROKS than TDDFT. The central claim is that ROKS achieves TDDFT-comparable accuracy at ground-state DFT cost for extended systems.
Significance. If the implementation and validations hold, the work is significant for enabling affordable excited-state simulations (including forces) in periodic systems with defects, where TDDFT scaling is often prohibitive. Explicit cross-code validation on molecules, direct comparison to TDDFT on a solid-state example, and functional-dependence tests provide concrete grounding. The derivation of analytical forces is a notable strength for geometry optimization and dynamics applications.
minor comments (3)
- [Abstract] Abstract: the unit is written as 'amu^{1/2} Ang'; standardize to 'amu^{1/2} Å' for consistency with standard notation in the field.
- [MgO application section] The manuscript should include a brief statement on the specific PAW potentials and plane-wave cutoff used for the MgO calculations to allow direct reproduction of the reported 0.21 eV deviation.
- [Implementation section] Clarify in the methods whether the ROKS singlet energy is obtained from the same weighted combination formula as in the Gaussian-basis reference implementation, or if any adjustment was made for the PAW representation.
Simulated Author's Rebuttal
We thank the referee for the positive and constructive review of our manuscript on the ROKS implementation in VASP. The recommendation for minor revision is noted, and we will incorporate appropriate updates in the revised version.
Circularity Check
No significant circularity; derivation self-contained via external benchmarks
full rationale
The paper's central claims rest on implementing ROKS within VASP's plane-wave PAW framework, deriving analytical forces, and validating via direct numerical comparison to the independent Q-Chem Gaussian-basis ROKS implementation (mean deviation ~30 meV across eight molecules) plus side-by-side comparison to TDDFT for MgO excitations (differences quantified at 0.21 eV, 0.14 eV, 0.12 amu^{1/2} Å). No equations, fitted parameters, or self-citations are shown that reduce reported energies or forces to the inputs by construction; the derivation chain is externally grounded rather than tautological.
Axiom & Free-Parameter Ledger
axioms (1)
- domain assumption The weighted mixed-spin plus triplet functional yields a spin-pure singlet when minimized
Reference graph
Works this paper leans on
-
[1]
(10) (Eq
However, we note that the stationarity condition Eq. (10) (Eq. 112 in Ref [32]) erroneously contained ⟨n|gm⟩ instead of ⟨gm|n⟩. Furthermore, it was stated that this condition is necessary if unitary transformations are allowed, while it is necessary ifnon-unitary transformations are allowed (see SI section S1). Nevertheless, the final definition ofγreported in
-
[2]
Using this definition ofγ, the gradient can be written as |gn⟩ = ∑ σ,L cL " f σL n 1− ∑ m S |m⟩ ⟨m| HσL |n⟩ | {z } :=|gnI⟩ +∑ m 1 2 (f σL n −f σL m )HσL mn S |m⟩ | {z } :=|gnII⟩ #
agrees with the definition derived in the present study. Using this definition ofγ, the gradient can be written as |gn⟩ = ∑ σ,L cL " f σL n 1− ∑ m S |m⟩ ⟨m| HσL |n⟩ | {z } :=|gnI⟩ +∑ m 1 2 (f σL n −f σL m )HσL mn S |m⟩ | {z } :=|gnII⟩ # . (12) The first contribution, |gnI⟩, represents the component of the gradient that lies outside the subspace spanned by...
-
[3]
This is identical to the variational conditions for the restricted open shell singlet derived in Refs [22, 42]. In Refs [22, 42], a DIIS method [43] was used to minimize the gradient in Eq. (26). We have implemented this DIIS solver as well as described in Ref [43] in a development version of V ASP. The implementation details will be reported elsewhere. T...
-
[4]
J. C. Slater, Statistical Exchange-Correlation in the Self- Consistent Field (Academic Press, 1972) pp. 1–92
1972
-
[5]
Ziegler, A
T. Ziegler, A. Rank, and E. J. Baerends, On the Calculation of Multiplet Energies by the Hartree-Fock-Slater Method, Theor. Chim. Acta43, 261 (1977)
1977
-
[6]
Gavnholt, T
J. Gavnholt, T. Olsen, M. Engelund, and J. Schiøtz,∆Self- Consistent Field Method to Obtain Potential Energy Surfaces of Excited Molecules on Surfaces, Phys. Rev. B78, 075441 (2008)
2008
-
[7]
G. Levi, A. V . Ivanov, and H. Jónsson, Variational Calculations of Excited States: Via Direct Optimization of the Orbitals in DFT, Faraday Discuss.224, 448 (2020)
2020
-
[8]
Hait and M
D. Hait and M. Head-Gordon, Orbital Optimized Density Func- tional Theory for Electronic Excited States, J. Phys. Chem. Lett. 12, 4517 (2021)
2021
-
[9]
A. Gali, M. Fyta, and E. Kaxiras, Ab Initio Supercell Calcu- lations on Nitrogen-Vacancy Center in Diamond: Electronic Structure and Hyperfine Tensors, Phys. Rev. B77, 155206 (2008)
2008
-
[10]
Alkauskas, B
A. Alkauskas, B. B. Buckley, D. D. Awschalom, and C. G. Van de Walle, First-Principles Theory of the Luminescence Lineshape for the Triplet Transition in Diamond NV Centres, New J. Phys.16, 073026 (2014)
2014
-
[11]
A. V . Ivanov, Y . L. Schmerwitch, G. Levi, and H. Jónsson, Elec- tronic Excitations of the Charged Nitrogen-Vacancy Center in Diamond Obtained using Time-Independent Variational Den- sity Functional Calculations, SciPost Phys.15, 10.21468/Sci- PostPhys.15.1.009 (2023)
-
[12]
Csóré, H
A. Csóré, H. J. von Bardeleben, J. L. Cantin, and A. Gali, Char- acterization and Formation of NV Centers in 3C,4H, And 6H SiC: An Ab Initio Study, Phys. Rev. B96, 085204 (2017)
2017
-
[13]
Mohseni, P
M. Mohseni, P. Udvarhelyi, G. m. H. Thiering, and A. Gali, Positively Charged Carbon Vacancy Defect as a Near-Infrared Emitter in 4H-SiC, Phys. Rev. Mater.7, 096202 (2023)
2023
-
[14]
Aberl, E
J. Aberl, E. P. Navarrete, M. Karaman, D. H. Enriquez, C. Wil- flingseder, A. Salomon, D. Primetzhofer, M. A. Schubert, G. Capellini, T. Fromherz, P. Deák, P. Udvarhelyi, S. Li, A. Gali, and M. Brehm, All-Epitaxial Self-Assembly of Silicon Color Centers Confined within Sub-Nanometer Thin Layers us- ing Ultra-Low Temperature Epitaxy, Adv. Mater.36, 2408424 (2024)
2024
-
[15]
M. E. Turiansky, A. Alkauskas, L. C. Bassett, and C. G. Van de Walle, Dangling Bonds in Hexagonal Boron Nitride as Single- Photon Emitters, Phys. Rev. Lett.123, 127401 (2019)
2019
-
[16]
Cholsuk, A
C. Cholsuk, A. Zand, A. Çakan, and T. V ogl, The hBN De- fects Database: A Theoretical Compilation of Color Centers in Hexagonal Boron Nitride, J. Phys. Chem. C128, 12716 (2024)
2024
-
[17]
A. Gale, M. Kianinia, J. Horder, C. Tweedie, M. Singhal, D. Scognamiglio, J. Qi, K. Liu, C. Verdi, I. Aharonovich, and M. Toth, Quantum Emitters in Rhombohedral Boron Nitride, Adv. Opt. Mater.13, e00593 (2025)
2025
-
[18]
A. T. B. Gilbert, N. A. Besley, and P. M. W. Gill, Self- Consistent Field Calculations of Excited States using the Max- imum Overlap Method (MOM), J. Phys. Chem. A112, 13164 (2008)
2008
-
[19]
G. M. J. Barca, A. T. B. Gilbert, and P. M. W. Gill, Simple Models for Difficult Electronic Excitations, J. Chem. Theory Comput.14, 1501 (2018)
2018
-
[20]
Carter-Fenk and J
K. Carter-Fenk and J. M. Herbert, State-Targeted Energy Pro- jection: A Simple and Robust Approach to Orbital Relaxation of Non-Aufbau Self-Consistent Field Solutions, J. Chem. The- ory Comput.16, 5067 (2020)
2020
-
[22]
Y . L. Schmerwitz, G. Levi, and H. Jónsson, Calculations of Ex- cited Electronic States by Converging on Saddle Points using Generalized Mode Following, J. Chem. Theory Comput.19, 3634 (2023)
2023
-
[23]
M. B. Robin,Higher Excited States of Polyatomic Molecules, V ol. 3 (Academic, New York, 1985)
1985
-
[24]
Filatov and S
M. Filatov and S. Shaik, Spin-Restricted Density Functional Approach to the Open-Shell Problem, Chem. Phys. Lett.288, 689 (1998)
1998
-
[25]
Frank, J
I. Frank, J. Hutter, D. Marx, and M. Parrinello, Molecular Dy- namics in Low-Spin Excited States, J. Chem. Phys.108, 4060 (1998)
1998
-
[26]
Filatov and S
M. Filatov and S. Shaik, Application of Spin-Restricted Open- Shell Kohn-Sham Method to Atomic and Molecular Multiplet States, J. Chem. Phys.110, 116 (1999)
1999
-
[27]
R. Büchel, L. Álvarez, J. Grage, D. Maniscalco, and I. Frank, On the Simulation of Photoreactions using Re- stricted Open-Shell Kohn–Sham Theory, Molecules29, 10.3390/molecules29184509 (2024)
-
[28]
I. D. Fedorov, N. D. Orekhov, and V . V . Stegailov, Nonadi- abatic Effects and Excitonlike States During the Insulator-to- Metal Transition in Warm Dense Hydrogen, Phys. Rev. B101, 12 100101 (2020)
2020
-
[29]
Schwermann and N
C. Schwermann and N. L. Doltsinis, Exciton Transfer Free En- ergy from Car–Parrinello Molecular Dynamics, Phys. Chem. Chem. Phys.22, 10526 (2020)
2020
-
[30]
C. O. Diarra, M. Boero, E. Steveler, T. Heiser, and E. Mar- tin, Exciton Diffusion in Poly(3-Hexylthiophene) by First- Principles Molecular Dynamics, Phys. Chem. Chem. Phys.25, 15539 (2023)
2023
-
[31]
N. A. Fominykh and V . V . Stegailov, Exciton Diffusion in MoS2 Monolayer from First-Principles Molecular Dynamics, J. Chem. Phys.163, 114704 (2025)
2025
-
[32]
T. Kowalczyk, T. Tsuchimochi, P. T. Chen, L. Top, and T. V . V oorhis, Excitation Energies and Stokes Shifts from a Re- stricted Open-Shell Kohn-Sham Approach, J. Chem. Phys.138, 10.1063/1.4801790 (2013)
-
[33]
Hirao and H
K. Hirao and H. Nakatsuji, General SCF Operator Satisfying Correct Variational Condition, J. Chem. Phys.59, 1457 (1973)
1973
-
[34]
C. C. J. Roothaan, Self-Consistent Field Theory for Open Shells of Electronic Systems, Rev. Mod. Phys.32, 179 (1960)
1960
-
[35]
Kresse and J
G. Kresse and J. Furthmüller, Efficiency of Ab-Initio Total En- ergy Calculations for Metals and Semiconductors using a Plane- Wave Basis Set, Comput. Mater. Sci.6, 15 (1996)
1996
-
[36]
Kresse and D
G. Kresse and D. Joubert, From Ultrasoft Pseudopotentials to the Projector Augmented-Wave Method, Phys. Rev. B59, 1758 (1999)
1999
-
[37]
Kresse and J
G. Kresse and J. Furthmüller, Efficient Iterative Schemes for Ab Initio Total-Energy Calculations using a Plane-Wave Basis Set, Phys. Rev. B54, 11169 (1996)
1996
-
[38]
P. E. Blöchl, Projector Augmented-Wave Method, Phys. Rev. B 50, 17953 (1994)
1994
-
[39]
Y . Shao, Z. Gan, E. Epifanovsky, A. T. Gilbert, M. Wormit, J. Kussmann, A. W. Lange, A. Behn, J. Deng, X. Feng,et al., Advances in Molecular Quantum Chemistry Contained in the Q-Chem 4 Program Package, Mol. Phys.113, 184 (2015)
2015
-
[40]
Epifanovsky, A
E. Epifanovsky, A. T. Gilbert, X. Feng, J. Lee, Y . Mao, N. Mardirossian, P. Pokhilko, A. F. White, M. P. Coons, A. L. Dempwolff,et al., Software for the Frontiers of Quantum Chemistry: An Overview of Developments in the Q-Chem 5 Package, J. Chem. Phys.155(2021)
2021
-
[41]
Fletcher and C
R. Fletcher and C. M. Reeves, Function Minimization by Con- jugate Gradients, Comput. J.7, 149 (1964)
1964
-
[42]
M. P. Teter, M. C. Payne, and D. C. Allan, Solution of Schrödinger’s Equation for Large Systems, Phys. Rev. B40, 12255 (1989)
1989
-
[43]
Jacobi, Über ein leichtes Verfahren die in der Theorie der Säcularstörungen vorkommenden Gleichungen numerisch aufzulösen*)., J
C. Jacobi, Über ein leichtes Verfahren die in der Theorie der Säcularstörungen vorkommenden Gleichungen numerisch aufzulösen*)., J. Reine Angew. Math.1846, 51 (1846)
-
[44]
W. D. Edwards and M. C. Zerner, A Generalized Restricted Open-Shell Fock Operator*, Theor. Chim. Acta72, 347 (1987)
1987
-
[45]
Grimm, C
S. Grimm, C. Nonnenberg, and I. Frank, Restricted Open- Shell Kohn-Sham Theory forφ−π ∗ Transitions. I. Polyenes, Cyanines, and Protonated Imines, J. Chem. Phys.119, 11574 (2003)
2003
-
[46]
Hutter, H
J. Hutter, H. P. Lüthi, and M. Parrinello, Electronic Structure Optimization in Plane-Wave-Based Density Functional Calcu- lations by Direct Inversion in the Iterative Subspace, Comput. Mater. Sci.2, 244 (1994)
1994
-
[47]
Carbó and J
R. Carbó and J. M. Riera,A General SCF Theory, Lecture Notes in Chemistry, V ol. 5 (Springer Berlin, Heidelberg, 1978)
1978
-
[48]
Friedrichs, K
J. Friedrichs, K. Damianos, and I. Frank, Solving Restricted Open-Shell Equations in Excited State Molecular Dynamics Simulations, Chem. Phys.347, 17 (2008), ultrafast Photoin- duced Processes in Polyatomic Molecules
2008
-
[49]
Goedecker and K
S. Goedecker and K. Maschke, Operator Approach in the Lin- earized Augmented-Plane-Wave Method: Efficient Electronic- Structure Calculations Including Forces, Phys. Rev. B45, 15 (1992)
1992
-
[50]
Hait and M
D. Hait and M. Head-Gordon, Excited State Orbital Optimiza- tion via Minimizing the Square of the Gradient: General Ap- proach and Application to Singly and Doubly Excited States via Density Functional Theory, J. Chem. Theory Comput.16, 1699 (2020)
2020
-
[51]
J. H. Skone, M. Govoni, and G. Galli, Self-Consistent Hybrid Functional for Condensed Systems, Phys. Rev. B89, 195112 (2014)
2014
-
[52]
Paier, R
J. Paier, R. Hirschl, M. Marsman, and G. Kresse, The Perdew–Burke–Ernzerhof Exchange-Correlation Functional Applied to the G2-1 Test Set using a Plane-Wave Basis Set, J. Chem. Phys.122, 234102 (2005)
2005
-
[53]
Rossomme, L
E. Rossomme, L. A. Cunha, W. Li, K. Chen, A. R. McIsaac, T. Head-Gordon, and M. Head-Gordon, The Good, the Bad, and the Ugly: Pseudopotential Inconsistency Errors in Molecular Applications of Density Functional Theory, J. Chem. Theory Comput.19, 2827 (2023)
2023
-
[54]
Y . Jin, V . W.-z. Yu, M. Govoni, A. C. Xu, and G. Galli, Excited State Properties of Point Defects in Semiconductors and In- sulators Investigated with Time-Dependent Density Functional Theory, J. Chem. Theory Comput.19, 8689 (2023)
2023
-
[55]
F. A. Evangelista, P. Shushkov, and J. C. Tully, Orthogonality Constrained Density Functional Theory for Electronic Excited States, J. Phys. Chem. A117, 7378 (2013)
2013
-
[56]
H. D. M. Pham and R. Z. Khaliullin, Direct Unconstrained Op- timization of Excited States in Density Functional Theory, J. Chem. Theory Comput.21, 3902 (2025)
2025
-
[57]
C. V orwerk and G. Galli, Disentangling Photoexcitation and Photoluminescence Processes in Defective MgO, Phys. Rev. Mater.7, 10.1103/PhysRevMaterials.7.033801 (2023)
-
[58]
D. J. Tozer and N. C. Handy, On the Determination of Exci- tation Energies using Density Functional Theory, Phys. Chem. Chem. Phys.2, 2117 (2000)
2000
-
[59]
K. Yang, R. Peverati, D. G. Truhlar, and R. Valero, Density Functional Study of Multiplicity-Changing Valence and Ryd- berg Excitations of P-Block Elements: Delta Self-Consistent Field, Collinear Spin-Flip Time-Dependent Density Functional Theory (DFT), and Conventional Time-Dependent DFT, J. Chem. Phys.135, 044118 (2011)
2011
-
[60]
Seidu, M
I. Seidu, M. Krykunov, and T. Ziegler, Applications of Time- Dependent and Time-Independent Density Functional Theory to Rydberg Transitions, J. Phys. Chem. A119, 5107 (2015)
2015
-
[61]
Liang, X
J. Liang, X. Feng, D. Hait, and M. Head-Gordon, Revisiting the Performance of Time-Dependent Density Functional The- ory for Electronic Excitations: Assessment of 43 Popular and Recently Developed Functionals from Rungs One to Four, J. Chem. Theory Comput.18, 3460 (2022)
2022
-
[62]
T. Kowalczyk, S. R. Yost, and T. V . V oorhis, Assessment of the∆-SCF Density Functional Theory Approach for Elec- tronic Excitations in Organic Dyes, J. Chem. Phys.134, 10.1063/1.3530801 (2011)
-
[63]
S. B. Worster, O. Feighan, and F. R. Manby, Reliable Transi- tion Properties from Excited-State Mean-Field Calculations, J. Chem. Phys.154, 10.1063/5.0041233 (2021)
-
[64]
is needed
R. Ramakrishnan, P. O. Dral, M. Rupp, and O. A. von Lilien- feld, Big Data Meets Quantum Chemistry Approximations: The∆-Machine Learning Approach, J. Chem. Theory Comput. 11, 2087 (2015). Supplementary Information: Excited States from Restricted Open Shell Plane-Wave DFT Michael J. Sahre* University of Vienna, Faculty of Physics, Kolingasse 14, A-1090 Vie...
2087
-
[65]
Hirao and H
K. Hirao and H. Nakatsuji, J. Chem. Phys.59, 1457 (1973)
1973
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.