DFT Accuracy on Crystal Structure Prediction with Machine Learning Interatomic Potentials
Pith reviewed 2026-06-29 09:23 UTC · model grok-4.3
The pith
CSP-MACE-Å reaches PBE and B86bPBE-XDM accuracy levels for ranking crystal structures on two separate test collections.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
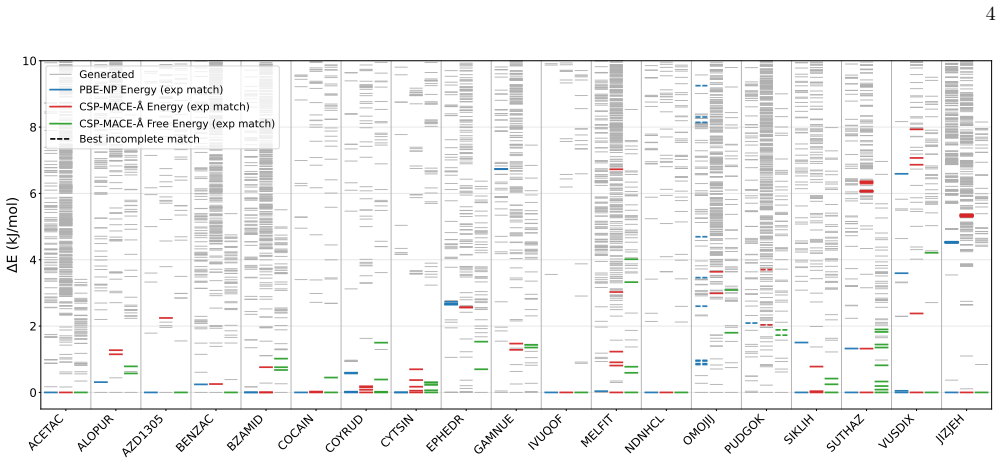
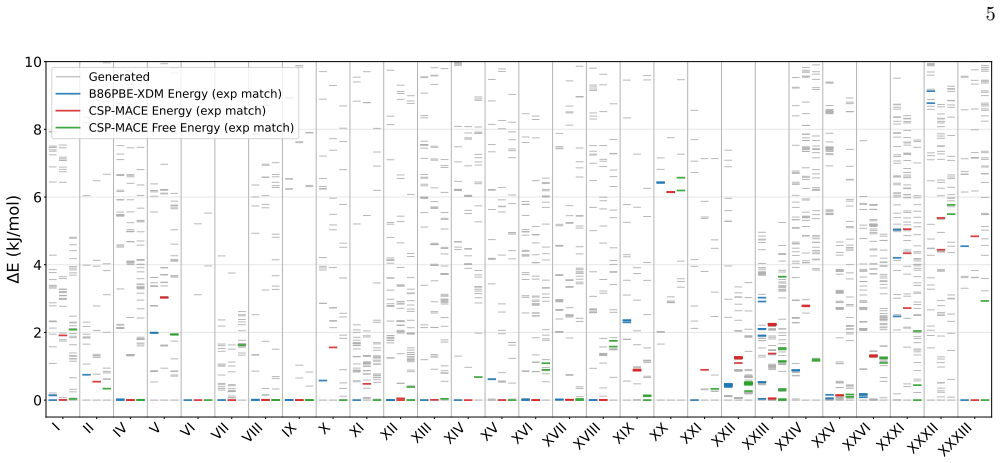
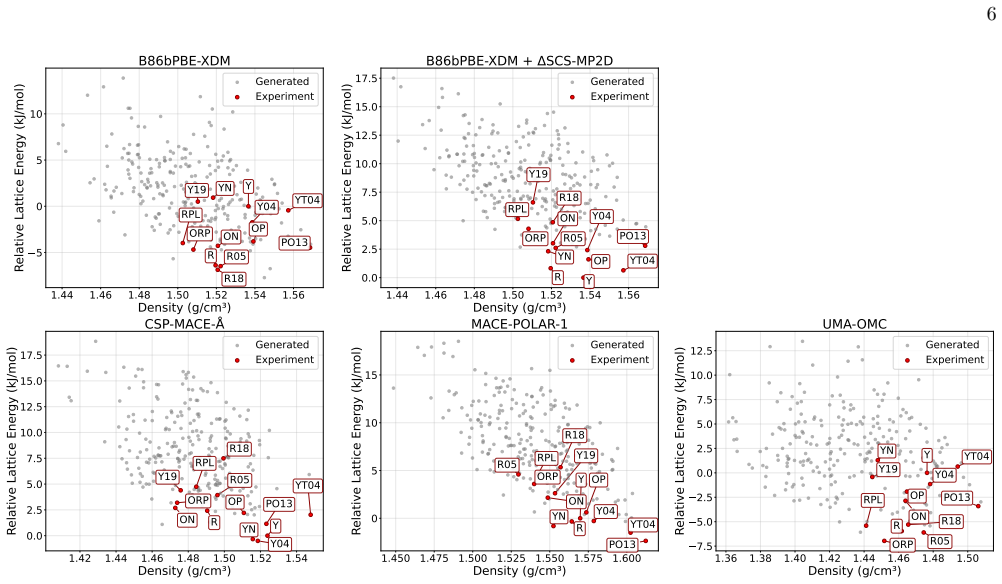
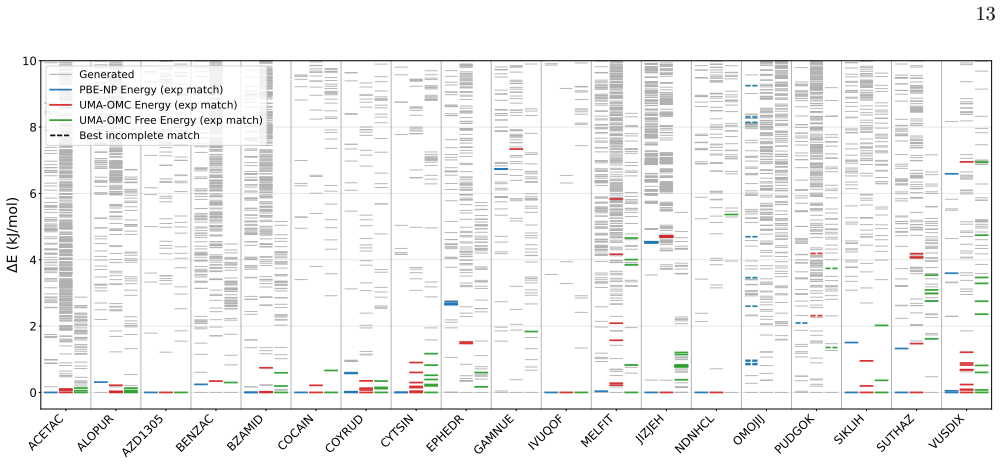
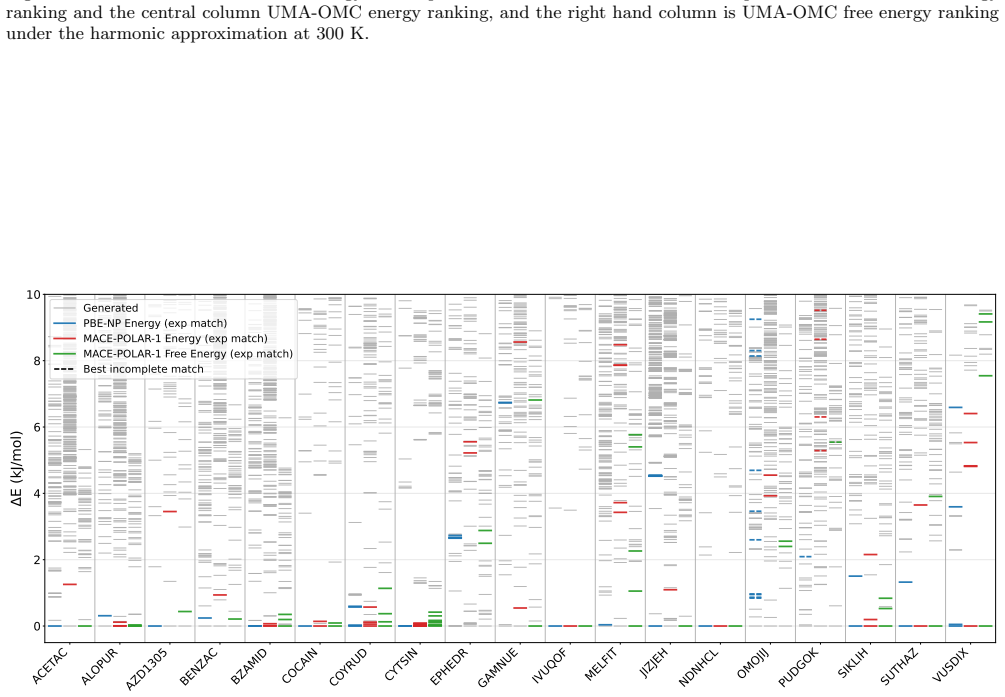
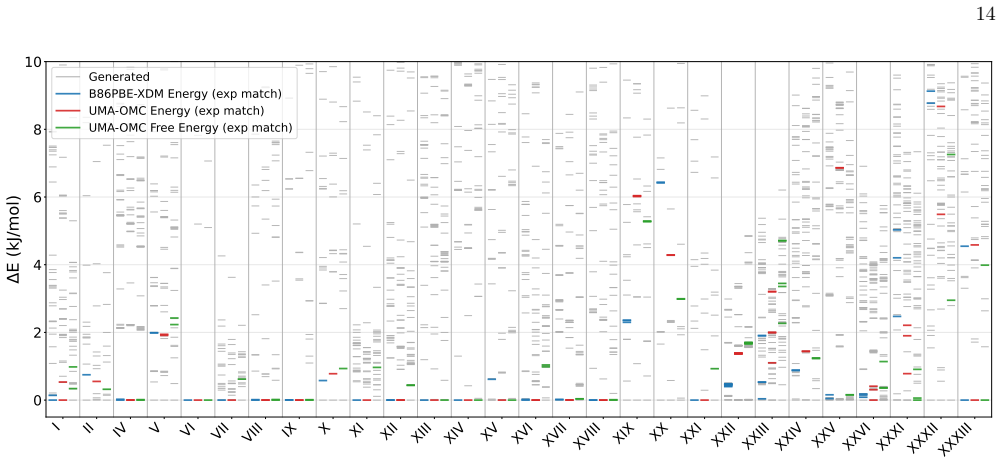
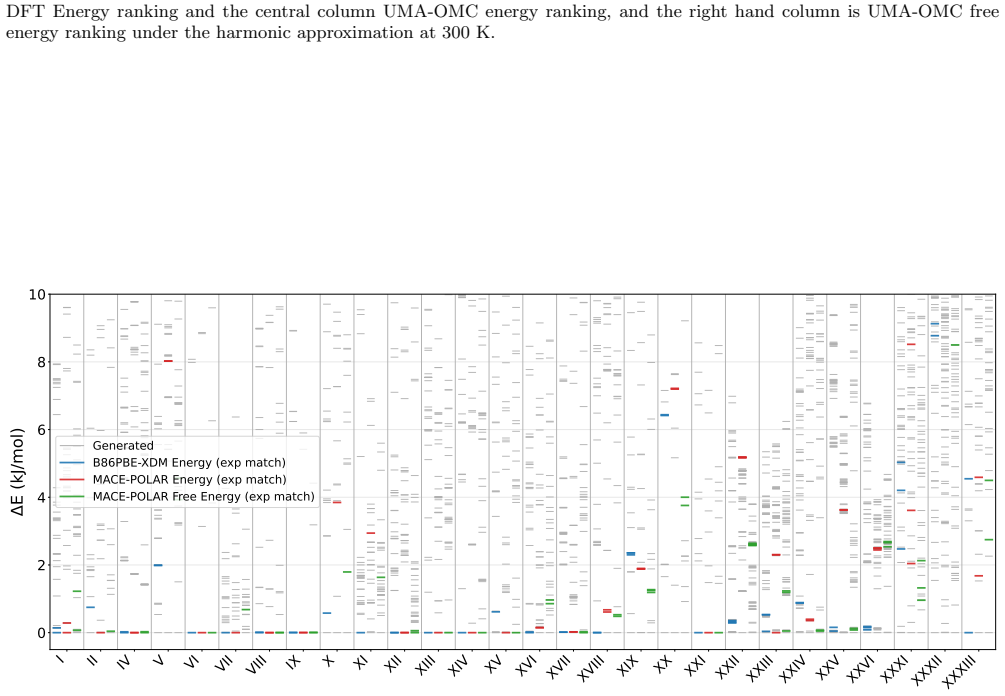
CSP-MACE-Å achieves performance comparable to PBE DFT with the Neumann-Perrin dispersion correction on a 19-compound AstraZeneca set and performance close to B86bPBE-XDM DFT on a 28-compound blind-test set that includes cocrystals and salts. Across both sets it outperforms the MACE-POLAR-1 and UMA-OMC foundation models, and on five compounds it reproduces temperature-dependent polymorph stability ordering when free energies are estimated under the harmonic approximation.
What carries the argument
CSP-MACE-Å, an energy decomposition that combines MACE-POLAR intramolecular and intermolecular contributions with an XDM-form dispersion term and a learned delta model trained on B86bPBE-XDM residuals from 50,000 structures.
If this is right
- Reranking candidate structures by harmonic free energy improves ranking accuracy over energy-only ranking on both evaluation sets.
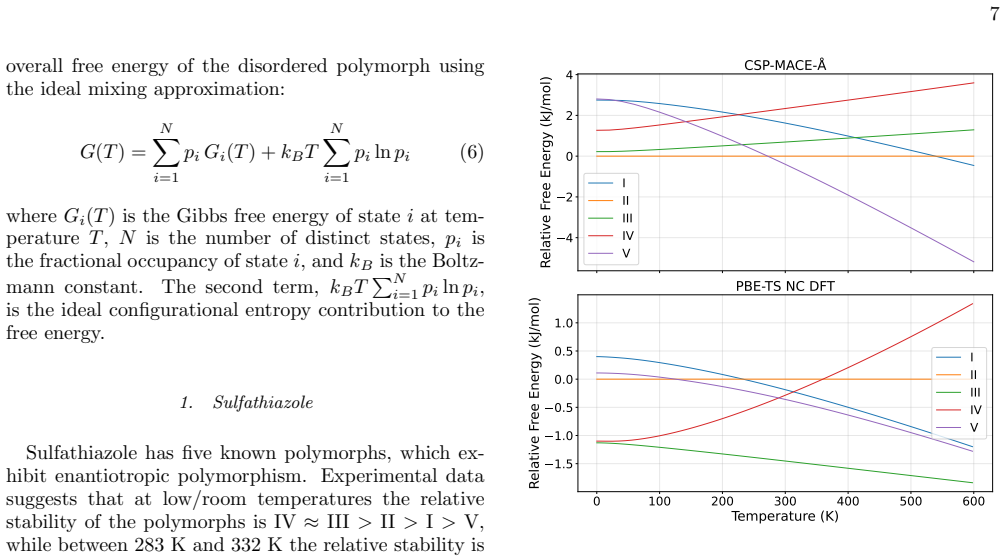
- The model reproduces temperature-dependent relative stability trends for five polymorph pairs.
- CSP-MACE-Å outperforms the MACE-POLAR-1 and UMA-OMC foundation models on the same test compounds.
- Because the model evaluates structures orders of magnitude faster than DFT, many more candidates can be scored in a single CSP campaign.
Where Pith is reading between the lines
- Wider adoption could allow routine inclusion of free-energy corrections in early-stage solid-form screening without added DFT cost.
- The decomposition into separate intra- and intermolecular models may transfer to other molecular crystal tasks such as solubility or nucleation modeling.
- Maintaining accuracy on salts and cocrystals already included in the tests suggests the approach can handle charged and multi-component systems once the delta model is retrained on appropriate residuals.
Load-bearing premise
The two evaluation collections of 19 and 28 compounds are representative of the chemical and structural variety found in actual crystal structure prediction work and do not overlap with the delta-model training data.
What would settle it
A fresh collection of crystal structures whose energy ordering under CSP-MACE-Å differs markedly from the ordering obtained with the reference DFT methods on the same structures would falsify the claim of comparable performance.
Figures
read the original abstract
We present an evaluation of CSP-MACE-{\AA}, a machine learning interatomic potential intended to replace DFT in crystal structure prediction (CSP). We decompose the total energy into separate intramolecular and intermolecular components. For the intramolecular component, we adopt the MACE-POLAR architecture and train it on the OMol25 dataset. The intermolecular component combines three terms: an intermolecular contribution from the MACE-POLAR model, a long-range dispersion term with the functional form of the XDM correction, and a learned delta model trained to reproduce B86bPBE-XDM intermolecular energies. The learned delta model is trained on residual intermolecular targets derived from 50,000 B86bPBE-XDM calculations on molecular crystal structures. On an evaluation set composed of 19 compounds, including a salt, selected from AstraZeneca's previous CSP publications, CSP-MACE-{\AA} achieves performance comparable to PBE DFT with the Neumann-Perrin dispersion correction. On a second evaluation set composed of 28 compounds, including cocrystals and salts, collated from the seven CSP blind tests, CSP-MACE-{\AA} achieves performance close to B86bPBE-XDM DFT. In both evaluation sets, reranking with harmonic free energies substantially improves performance relative to ranking by energy alone. Across our evaluation suite, CSP-MACE-{\AA} is shown to outperform the MACE-POLAR-1 and UMA-OMC foundation models. Lastly, on a set of five compounds, CSP-MACE-{\AA} is shown to capture temperature-dependent trends in the relative stability of polymorphs through estimation of the free energy under the harmonic approximation. By running multiple orders of magnitude faster than DFT, CSP-MACE-{\AA} enables energy and free energy evaluation of far more candidate structures, providing greater confidence when derisking solid forms.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript presents CSP-MACE-Å, an ML interatomic potential for crystal structure prediction that decomposes energy into intramolecular (MACE-POLAR trained on OMol25) and intermolecular (MACE-POLAR + XDM dispersion + learned delta model) components. The delta model is trained on residuals from 50,000 B86bPBE-XDM calculations. On a 19-compound AstraZeneca-derived set (including a salt), it claims performance comparable to PBE+Neumann-Perrin DFT; on a 28-compound set from CSP blind tests (including cocrystals and salts), performance close to B86bPBE-XDM DFT. Harmonic free-energy reranking improves results in both cases, and the model outperforms MACE-POLAR-1 and UMA-OMC while capturing temperature-dependent polymorph trends on five compounds via harmonic free energies.
Significance. If the evaluation sets are verifiably disjoint from the 50k training structures, the work would demonstrate a practical route to DFT-level accuracy in CSP at orders-of-magnitude lower cost, enabling exhaustive ranking of far larger candidate pools and greater confidence in solid-form selection. The energy decomposition and hybrid physical/ML intermolecular correction are technically interesting strengths.
major comments (2)
- [Abstract] Abstract: The manuscript provides no explicit statement, table, or supplementary note confirming that none of the 50,000 B86bPBE-XDM structures (or close structural analogues) used to train the learned delta model overlap with the 19 AstraZeneca or 28 blind-test evaluation compounds. This disjointness is load-bearing for the central generalization claim that CSP-MACE-Å matches DFT performance on independent sets.
- [Abstract] Abstract: No quantitative metrics (e.g., lattice RMSD, success rates at top-1/3/10, or confidence intervals) are reported for the claimed comparability to PBE+Neumann-Perrin or B86bPBE-XDM on the two evaluation sets, only qualitative statements. This prevents assessment of effect size and statistical robustness.
Simulated Author's Rebuttal
We thank the referee for their thoughtful review and for highlighting two important points regarding the abstract. We address each comment below and will incorporate revisions to improve clarity and rigor.
read point-by-point responses
-
Referee: [Abstract] Abstract: The manuscript provides no explicit statement, table, or supplementary note confirming that none of the 50,000 B86bPBE-XDM structures (or close structural analogues) used to train the learned delta model overlap with the 19 AstraZeneca or 28 blind-test evaluation compounds. This disjointness is load-bearing for the central generalization claim that CSP-MACE-Å matches DFT performance on independent sets.
Authors: We agree that explicit confirmation of disjointness is essential to support the generalization claims. The 19 AstraZeneca and 28 blind-test compounds were selected to be independent of the 50,000 training structures, with no structural overlap or close analogues (verified via fingerprint similarity and manual inspection during dataset curation). In the revised manuscript we will add an explicit statement to the abstract and include a supplementary note detailing the checks performed to confirm disjointness. revision: yes
-
Referee: [Abstract] Abstract: No quantitative metrics (e.g., lattice RMSD, success rates at top-1/3/10, or confidence intervals) are reported for the claimed comparability to PBE+Neumann-Perrin or B86bPBE-XDM on the two evaluation sets, only qualitative statements. This prevents assessment of effect size and statistical robustness.
Authors: We acknowledge that the abstract currently uses only qualitative phrasing. The main text and supplementary information contain the requested quantitative results, including top-1/3/10 success rates, lattice RMSD values, and comparisons across methods. To address the concern, we will revise the abstract to incorporate specific quantitative metrics (e.g., success rates and RMSDs) that quantify the comparability to the reference DFT methods. revision: yes
Circularity Check
No circularity: performance claims rest on generalization to external evaluation compounds
full rationale
The paper constructs CSP-MACE-Å by training a delta model on residuals from 50,000 B86bPBE-XDM calculations and then reports its performance on two separate evaluation sets (19 AstraZeneca compounds and 28 blind-test compounds) that are described as collated from prior external publications. These evaluation compounds are distinct from the training structures used for the delta model, making the reported comparability to PBE+Neumann-Perrin and B86bPBE-XDM an independent test of out-of-distribution accuracy rather than a definitional or fitted-input reduction. No self-citations, uniqueness theorems, or ansatzes are invoked as load-bearing steps in the provided text, and the central claims do not reduce to the inputs by construction.
Axiom & Free-Parameter Ledger
free parameters (1)
- learned delta model parameters
axioms (1)
- domain assumption Decomposition of total crystal energy into separate intramolecular and intermolecular components is sufficiently accurate for the systems studied.
Reference graph
Works this paper leans on
-
[1]
show that B86bPBE-XDM is inaccurate for ROY due to delocalisation error but this can be corrected by replacing the modelling of intramolecular interac- tions with spin-component-scaled dispersion-corrected second-order Møller–Plesset perturbation theory (SCS-MP2D) with an equivalent form to Equation 2. SCS-MP2D is a suitable choice because it has been sho...
2048
-
[2]
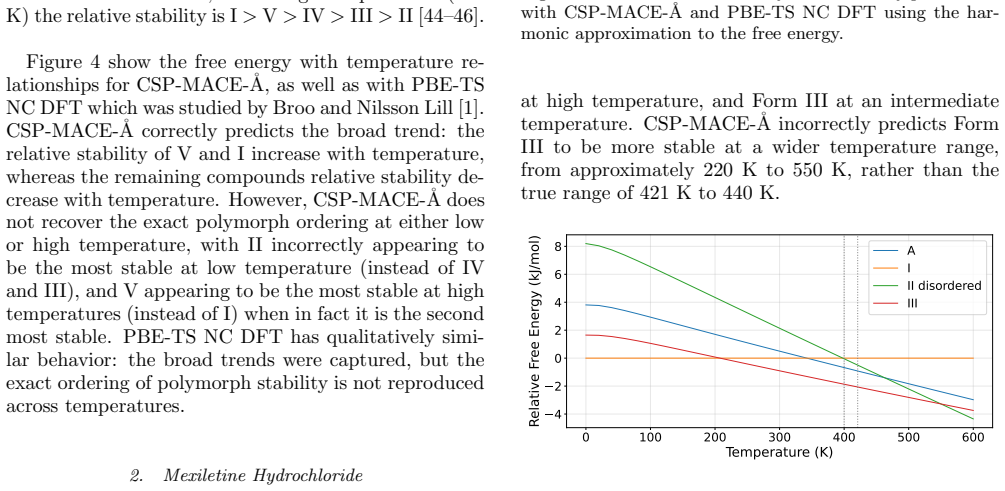
Sulfathiazole Sulfathiazole has five known polymorphs, which ex- hibit enantiotropic polymorphism. Experimental data suggests that at low/room temperatures the relative stability of the polymorphs is IV≈III>II>I>V, while between 283 K and 332 K the relative stability is III>II>IV>V>I , and at high temperatures (450 K) the relative stability is I>V>IV>III>...
-
[3]
Form I, II and III are anhydrous, and Types A and B are families of channel solvates with the addition of a nonsolvated form isostructural to the Type A solvate [26]
Mexiletine Hydrochloride Mexiletine hydrochloride is a salt with five solid-form types. Form I, II and III are anhydrous, and Types A and B are families of channel solvates with the addition of a nonsolvated form isostructural to the Type A solvate [26]. Form II has disorder with two distinct con- formations of the mexiletine molecule. Here we study the r...
-
[4]
AZD1305 AZD1305 is a crystalline oxabispidine pharmaceuti- cal with two polymorphs, Form A and B. Experimental data suggests the forms have a monotropic relationship, with Form B more stable at all temperatures, while their relative stability becomes closer at elevated tem- peratures [47]. Figure 6 shows that CSP-MACE- ˚A cor- 8 rectly predicts the monotr...
-
[5]
Experimental data suggests Form B is more stable at room temperature and Form A is more stable at high temperature
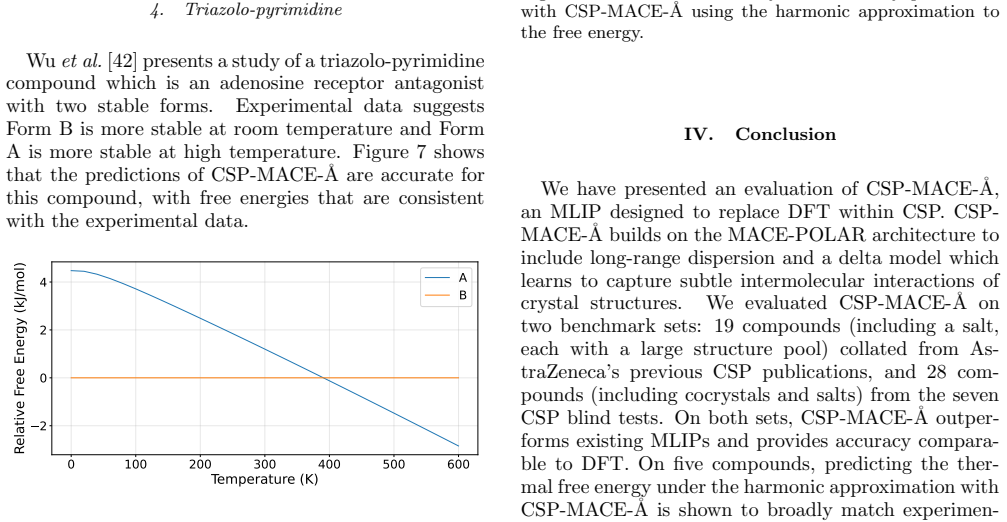
Triazolo-pyrimidine Wuet al.[42] presents a study of a triazolo-pyrimidine compound which is an adenosine receptor antagonist with two stable forms. Experimental data suggests Form B is more stable at room temperature and Form A is more stable at high temperature. Figure 7 shows that the predictions of CSP-MACE- ˚A are accurate for this compound, with fre...
-
[6]
It has two forms which exhibit enan- tiotropic polymorphism
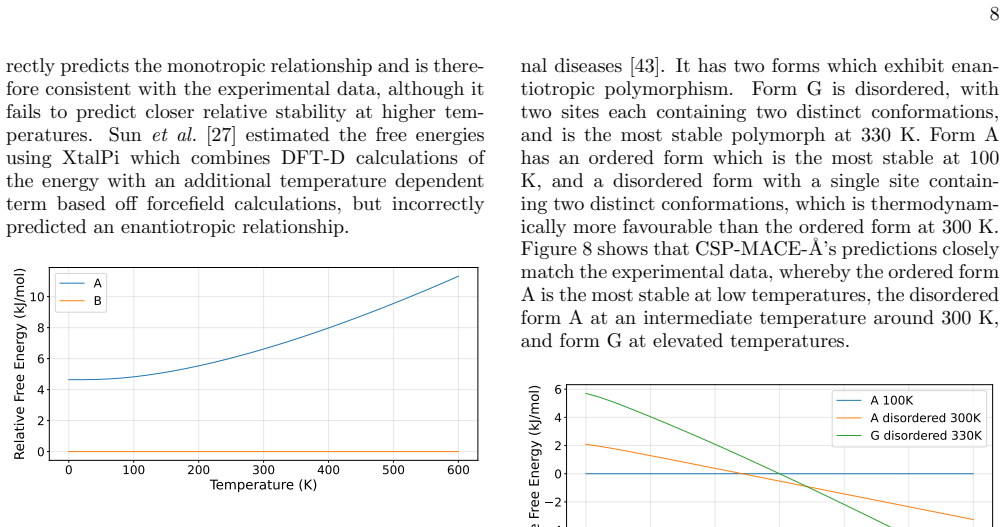
AZD5462 AZD5462 is an agonist of the relaxin family peptide receptor, being developed for the treatment of cardiore- nal diseases [43]. It has two forms which exhibit enan- tiotropic polymorphism. Form G is disordered, with two sites each containing two distinct conformations, and is the most stable polymorph at 330 K. Form A has an ordered form which is ...
-
[7]
Broo and S
A. Broo and S. O. Nilsson Lill, Transferable force field for crystal structure predictions, investigation of perfor- mance and exploration of different rescoring strategies using dft-d methods, Acta Crystallographica Section B 72, 460 (2016)
2016
-
[8]
J. P. Lommerse, W. S. Motherwell, H. L. Ammon, J. D. Dunitz, A. Gavezzotti, D. W. Hofmann, F. J. Leusen, W. T. Mooij, S. L. Price, B. Schweizer,et al., A test of crystal structure prediction of small organic molecules, Structural Science56, 697 (2000)
2000
-
[9]
W. S. Motherwell, H. L. Ammon, J. D. Dunitz, A. Dzyabchenko, P. Erk, A. Gavezzotti, D. W. Hof- mann, F. J. Leusen, J. P. Lommerse, W. T. Mooij,et al., Crystal structure prediction of small organic molecules: a second blind test, Structural Science58, 647 (2002)
2002
-
[10]
G. Day, W. Motherwell, H. Ammon, S. Boerrigter, R. G. Della Valle, E. Venuti, A. Dzyabchenko, J. D. Dunitz, B. Schweizer, B. Van Eijck,et al., A third blind test of crystal structure prediction, Structural Science 61, 511 (2005)
2005
-
[11]
G. M. Day, T. G. Cooper, A. J. Cruz-Cabeza, K. E. Hejczyk, H. L. Ammon, S. X. Boerrigter, J. S. Tan, R. G. Della Valle, E. Venuti, J. Jose,et al., Significant progress in predicting the crystal structures of small or- ganic molecules–a report on the fourth blind test, Struc- tural Science65, 107 (2009)
2009
-
[12]
D. A. Bardwell, C. S. Adjiman, Y. A. Arnautova, E. Bartashevich, S. X. Boerrigter, D. E. Braun, A. J. Cruz-Cabeza, G. M. Day, R. G. Della Valle, G. R. De- siraju,et al., Towards crystal structure prediction of complex organic compounds–a report on the fifth blind test, Structural Science67, 535 (2011)
2011
-
[13]
A. M. Reilly, R. I. Cooper, C. S. Adjiman, S. Bhat- tacharya, A. D. Boese, J. G. Brandenburg, P. J. By- grave, R. Bylsma, J. E. Campbell, R. Car,et al., Re- port on the sixth blind test of organic crystal structure prediction methods, Structural Science72, 439 (2016)
2016
-
[14]
L. M. Hunnisett, N. Francia, J. Nyman, N. S. Abraham, S. Aitipamula, T. Alkhidir, M. Almehairbi, A. Anelli, D. M. Anstine, J. E. Anthony,et al., The seventh blind test of crystal structure prediction: structure ranking methods, Structural Science80(2024)
2024
-
[15]
L. M. Hunnisett, J. Nyman, N. Francia, N. S. Abraham, C. S. Adjiman, S. Aitipamula, T. Alkhidir, M. Alme- hairbi, A. Anelli, D. M. Anstine,et al., The seventh blind test of crystal structure prediction: structure gen- eration methods, Structural Science80, 517 (2024)
2024
-
[16]
Firaha, Y
D. Firaha, Y. M. Liu, J. van de Streek, K. Sasiku- mar, H. Dietrich, J. Helfferich, L. Aerts, D. E. Braun, A. Broo, A. G. DiPasquale,et al., Predicting crystal form stability under real-world conditions, Nature623, 324 (2023)
2023
-
[17]
I. Batatia, W. J. Baldwin, D. Kuryla, J. Hart, E. Ka- soar, A. M. Elena, H. Moore, M. J. Gawkowski, B. X. Shi, V. Kapil, P. Kourtis, I.-B. Magd˘ au, and G. Cs´ anyi, Mace-polar-1: A polarisable electrostatic foundation model for molecular chemistry (2026), arXiv:2602.19411 [physics.chem-ph]
- [18]
-
[19]
C. J. Nickerson and E. R. Johnson, Assessment of a foundational machine-learned potential for energy rank- ing of molecular crystal polymorphs, Physical Chem- istry Chemical Physics27, 11930 (2025)
2025
-
[20]
Greenwell and G
C. Greenwell and G. J. Beran, Inaccurate conforma- tional energies still hinder crystal structure prediction in flexible organic molecules, Crystal Growth & Design 20, 4875 (2020)
2020
-
[21]
Mardirossian and M
N. Mardirossian and M. Head-Gordon,ωb97m-v: A combinatorially optimized, range-separated hybrid, meta-gga density functional with vv10 nonlocal corre- lation, The Journal of chemical physics144(2016)
2016
-
[22]
Rappoport and F
D. Rappoport and F. Furche, Property-optimized gaus- sian basis sets for molecular response calculations, The Journal of chemical physics133(2010)
2010
-
[23]
Hujo and S
W. Hujo and S. Grimme, Performance of the van der waals density functional vv10 and (hybrid) gga vari- ants for thermochemistry and noncovalent interactions, Journal of Chemical Theory and Computation7, 3866 (2011)
2011
-
[24]
Becke, On the large-gradient behavior of the den- sity functional exchange energy, The Journal of chemi- cal physics85, 7184 (1986)
A. Becke, On the large-gradient behavior of the den- sity functional exchange energy, The Journal of chemi- cal physics85, 7184 (1986)
1986
-
[25]
J. P. Perdew, K. Burke, and M. Ernzerhof, Generalized gradient approximation made simple, Physical review letters77, 3865 (1996)
1996
-
[26]
A. D. Becke and E. R. Johnson, Exchange-hole dipole moment and the dispersion interaction revisited, The Journal of chemical physics127(2007)
2007
-
[27]
E. R. Johnson, The exchange-hole dipole moment dis- persion model, Non-covalent interactions in quantum chemistry and physics , 169 (2017)
2017
-
[28]
A. J. Price, R. A. Mayo, A. Otero-de-la Roza, and E. R. Johnson, Accurate and efficient polymorph energy rank- ing with xdm-corrected hybrid dft, CrystEngComm25, 953 (2023)
2023
-
[29]
R. A. Mayo, A. J. Price, A. Otero-de-la Roza, and E. R. Johnson, Assessment of the exchange-hole dipole mo- ment dispersion correction for the energy ranking stage of the seventh crystal structure prediction blind test, Structural Science80(2024)
2024
-
[30]
Della Pia, B
F. Della Pia, B. X. Shi, V. Kapil, A. Zen, D. Alf` e, and A. Michaelides, Accurate and efficient machine learn- ing interatomic potentials for finite temperature mod- elling of molecular crystals, Chemical Science16, 11419 (2025). 10
2025
-
[31]
J. A. Chisholm and S. Motherwell, Compack: a pro- gram for identifying crystal structure similarity using distances, Applied Crystallography38, 228 (2005)
2005
-
[32]
J. L. Andrews, S. O. Nilsson Lill, S. Freitag-Pohl, D. C. Apperley, D. S. Yufit, A. S. Batsanov, M. T. Mulvee, K. Edkins, J. F. McCabe, D. J. Berry,et al., Derisking the polymorph landscape: the complex polymorphism of mexiletine hydrochloride, Crystal Growth & Design 21, 7150 (2021)
2021
-
[33]
G. Sun, X. Liu, Y. A. Abramov, S. O. Nilsson Lill, C. Chang, V. Burger, and A. Broo, Current state-of- the-art in-house and cloud-based applications of virtual polymorph screening of pharmaceutical compounds: a challenging case of azd1305, Crystal Growth & Design 21, 1972 (2021)
1972
-
[34]
M. A. Neumann, F. J. Leusen, and J. Kendrick, A ma- jor advance in crystal structure prediction, Angewandte Chemie International Edition47, 2427 (2008)
2008
-
[35]
Tkatchenko and M
A. Tkatchenko and M. Scheffler, Accurate molecular van der waals interactions from ground-state electron density¡? format?¿ and free-atom reference data, Phys- ical review letters102, 073005 (2009)
2009
-
[36]
M. A. Neumann and M.-A. Perrin, Energy ranking of molecular crystals using density functional theory cal- culations and an empirical van der waals correction, The Journal of Physical Chemistry B109, 15531 (2005)
2005
-
[37]
Gharakhanyan, L
V. Gharakhanyan, L. Barroso-Luque, Y. Yang, M. Shuaibi, K. Michel, D. S. Levine, M. Dzamba, X. Fu, M. Gao, X. Liu,et al., Open molecular crystals 2025 (omc25) dataset and models, Scientific Data (2026)
2025
-
[38]
V. Gharakhanyan, Y. Yang, L. Barroso-Luque, M. Shuaibi, D. S. Levine, K. Michel, V. Bernat, M. Dzamba, X. Fu, M. Gao,et al., Fastcsp: Acceler- ated molecular crystal structure prediction with univer- sal model for atoms, arXiv preprint arXiv:2508.02641 (2025)
-
[39]
Bitzek, P
E. Bitzek, P. Koskinen, F. G¨ ahler, M. Moseler, and P. Gumbsch, Structural relaxation made simple, Phys. Rev. Lett.97, 170201 (2006)
2006
-
[40]
C. G. Broyden, The convergence of a class of double- rank minimization algorithms 1. general considerations, IMA Journal of Applied Mathematics6, 76 (1970)
1970
-
[41]
Fletcher, A new approach to variable metric algo- rithms, The computer journal13, 317 (1970)
R. Fletcher, A new approach to variable metric algo- rithms, The computer journal13, 317 (1970)
1970
-
[42]
Goldfarb, A family of variable-metric methods de- rived by variational means, Mathematics of computa- tion24, 23 (1970)
D. Goldfarb, A family of variable-metric methods de- rived by variational means, Mathematics of computa- tion24, 23 (1970)
1970
-
[43]
D. F. Shanno, Conditioning of quasi-newton methods for function minimization, Mathematics of computation 24, 647 (1970)
1970
-
[44]
Otero-De-La-Roza, E
A. Otero-De-La-Roza, E. R. Johnson, and V. Lua˜ na, Critic2: A program for real-space analysis of quantum chemical interactions in solids, Computer Physics Com- munications185, 1007 (2014)
2014
-
[45]
Weatherston, M
J. Weatherston, M. R. Probert, and M. J. Hall, Poly- morphic royalty: the 14th roy polymorph discovered via high-throughput crystallization, Journal of the Ameri- can Chemical Society147, 11949 (2025)
2025
-
[46]
Nyman, L
J. Nyman, L. Yu, and S. M. Reutzel-Edens, Accuracy and reproducibility in crystal structure prediction: the curious case of roy, CrystEngComm21, 2080 (2019)
2080
-
[47]
G. J. Beran, I. J. Sugden, C. Greenwell, D. H. Bowskill, C. C. Pantelides, and C. S. Adjiman, How many more polymorphs of roy remain undiscovered, Chemical sci- ence13, 1288 (2022)
2022
-
[48]
D. Wu, E. S. Eriksson, S. O. N. Lill, J. F. McCabe, C. Bauer, and M. L. Lamb, Discovery of the most stable form of an adenosine receptor antagonist through vir- tual polymorph screening and targeted crystallization, Journal of Pharmaceutical Sciences114, 829 (2025)
2025
-
[49]
O. D. Putra, M. Lindhagen, J. Sjostrom, P. A. Cor- ner, E. M. Dodd, E. S. Eriksson, S. O. Nilsson Lill, and J. F. McCabe, Mechanistic understanding of solid– solid phase transition based on in situ single-crystal-to- single-crystal transformations: A case study of azd5462, Crystal Growth & Design25, 1565 (2025)
2025
-
[50]
M. M. Parmar, O. Khan, L. Seton, and J. L. Ford, Polymorph selection with morphology control using sol- vents, Crystal Growth & Design7, 1635 (2007)
2007
-
[51]
Munroe, ˚A
A. Munroe, ˚A. C. Rasmuson, B. K. Hodnett, and D. M. Croker, Relative stabilities of the five polymorphs of sulfathiazole, Crystal growth & design12, 2825 (2012)
2012
-
[52]
Sovago, M
I. Sovago, M. J. Gutmann, J. G. Hill, H. M. Senn, L. H. Thomas, C. C. Wilson, and L. J. Farrugia, Experimen- tal electron density and neutron diffraction studies on the polymorphs of sulfathiazole, Crystal growth & de- sign14, 1227 (2014)
2014
-
[53]
Sigfridsson, R
K. Sigfridsson, R. Lundqvist, and K. Ohlson, Preformu- lation evaluation of azd1305, an oxabispidine intended for oral and intravenous treatment, Drug development and industrial pharmacy38, 19 (2012). A. Appendix
2012
-
[54]
AZ Evaluation Set The number of structures from each stage of CSP on the AZ set is not consistent across compounds, as this set is collated from multiple CSP analyses
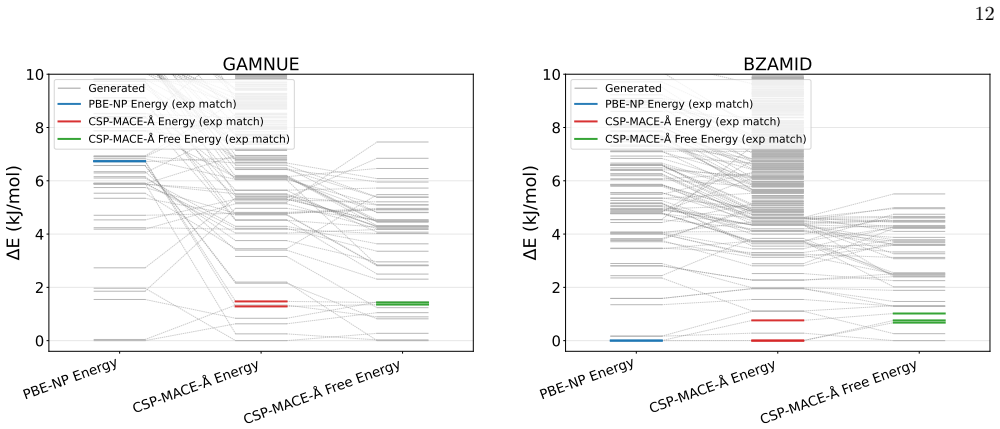
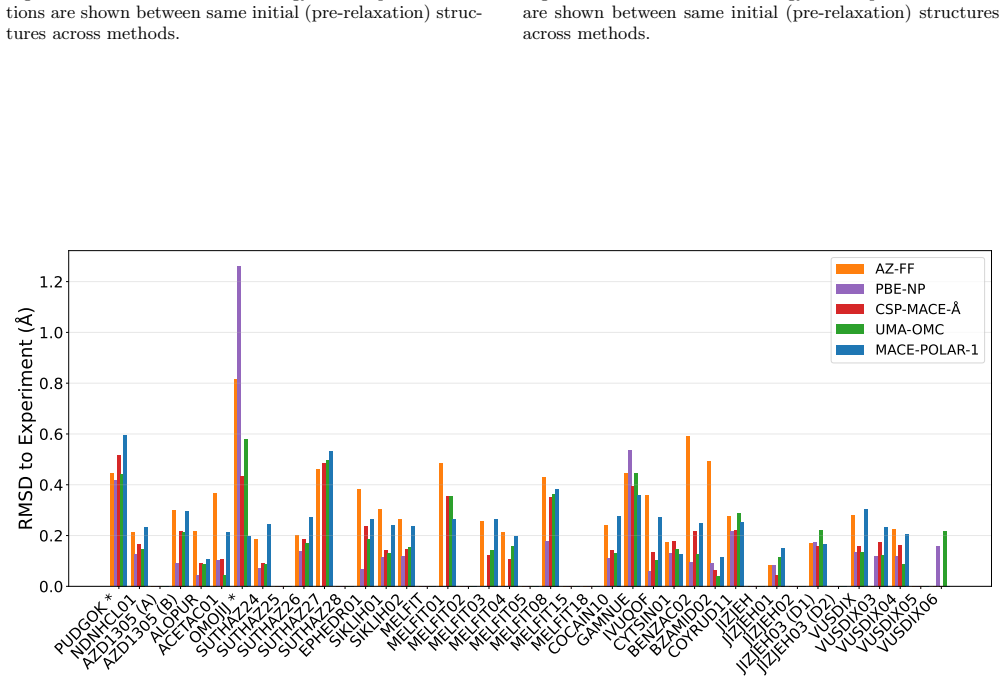
Experimental details and Further Results a. AZ Evaluation Set The number of structures from each stage of CSP on the AZ set is not consistent across compounds, as this set is collated from multiple CSP analyses. MLIPs reranking is applied to all available AZ-FF structures (typically around 1000 structures), while PBE-NP DFT relaxation is done on a smaller...
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.