A Triple-Modal Contrastive Learning Framework with Sequence, Graph, and 3D Features for Drug-Target Interaction Prediction
Pith reviewed 2026-06-29 08:39 UTC · model grok-4.3
The pith
TriMod-DTI combines sequence, graph, and 3D features through contrastive learning to improve drug-target interaction predictions.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
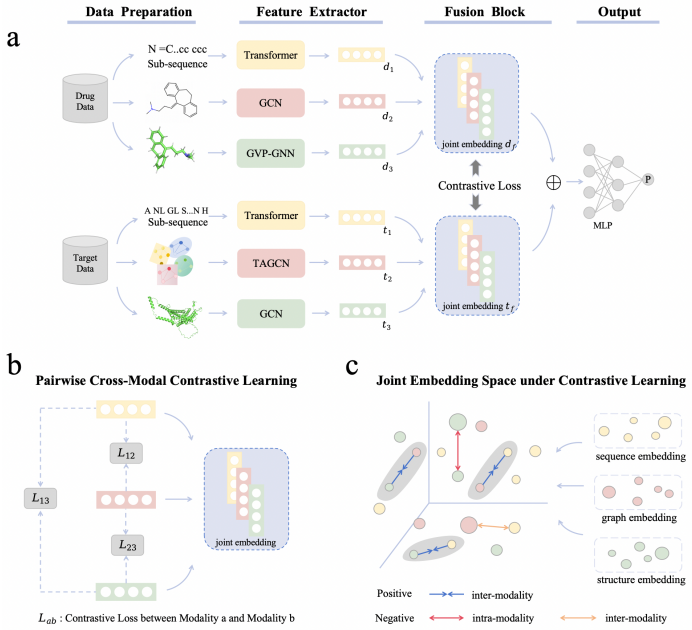
TriMod-DTI is a triple-modal contrastive learning framework that incorporates 1D sequences, 2D graphs, and 3D structures of drugs and proteins, obtaining the universal and complementary feature representations for DTI prediction. A Feature Extractor captures drug and target features across the three modalities. A triple-modal contrastive learning strategy aligns different modal representations of the same drug or protein in the latent space by constructing cross-modal positive and negative sample pairs, thereby enhancing the model's discriminative ability.
What carries the argument
The triple-modal contrastive learning strategy that aligns representations from sequence, graph, and 3D modalities of the same drug or protein through cross-modal positive and negative pairs.
If this is right
- The model outperforms state-of-the-art methods on three benchmark datasets for DTI prediction.
- Ablation studies confirm that each of the three modalities contributes to overall performance.
- Case studies demonstrate practical utility for identifying potential drug-target pairs in drug discovery workflows.
- The framework produces enriched representations by integrating information across 1D, 2D, and 3D views that prior single- or dual-modal approaches overlook.
Where Pith is reading between the lines
- The same alignment approach might transfer to other bioinformatics tasks that already have access to sequence, graph, and structural data, such as protein-protein interaction prediction.
- If the contrastive step reduces reliance on large labeled sets, follow-up work could test performance in low-data regimes typical of rare targets.
- Adding a fourth modality, such as textual descriptions from literature, could be tested as a direct extension of the current three-way alignment design.
Load-bearing premise
The three modalities supply complementary rather than redundant information and the contrastive alignment step meaningfully improves downstream DTI prediction accuracy.
What would settle it
Training the same model on the benchmark datasets after removing the contrastive alignment component and observing no drop in prediction metrics would falsify the contribution of the alignment step.
Figures


read the original abstract
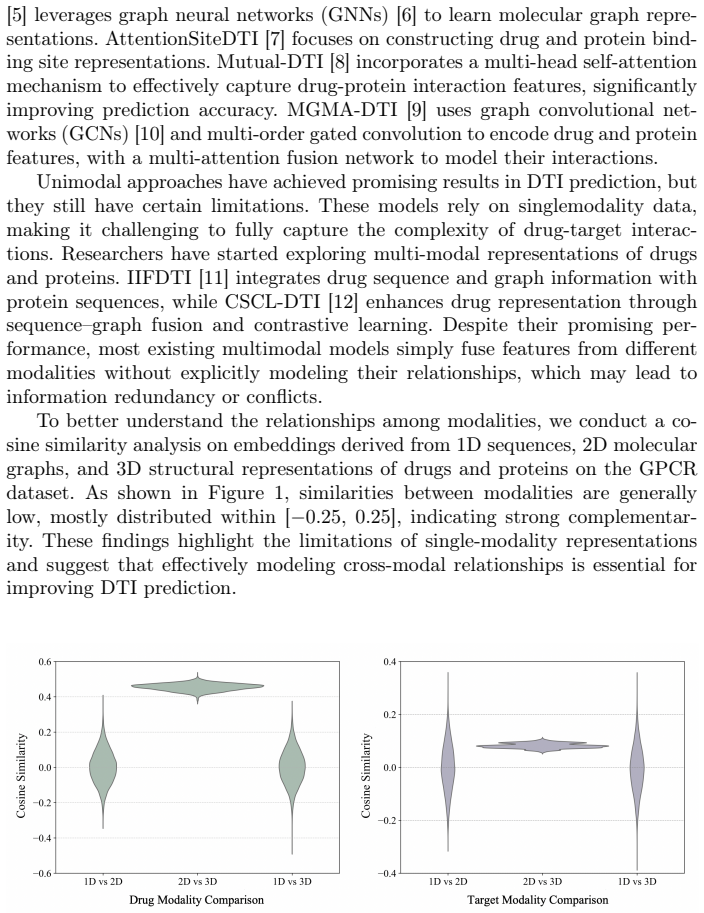
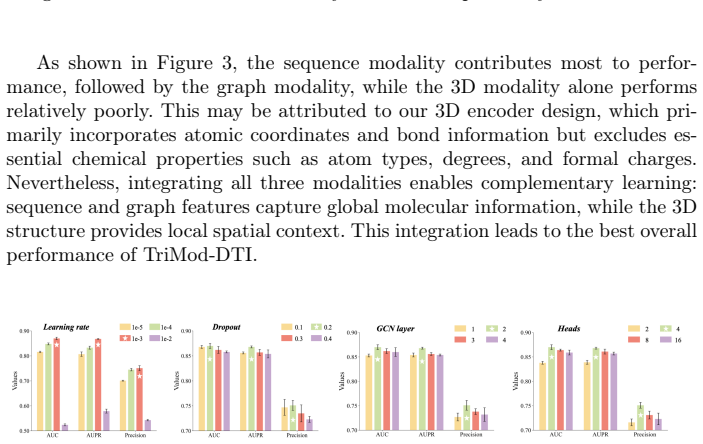
Accurate prediction of drug-target interactions (DTI) is critical for drug discovery. Existing methods often rely on single-modal representations (e.g., sequences or graphs) or combine only two modalities, overlooking 3D structural features. To address this challenge, we propose TriMod-DTI, a triple-modal contrastive learning framework that incorporates 1D sequences, 2D graphs, and 3D structures of drugs and proteins, obtaining the universal and complementary feature representations for DTI prediction. We design a Feature Extractor to capture drug and target features across the three modalities, thereby enriching their representations. We further propose a triple-modal contrastive learning strategy to align different modal representations of the same drug or protein in the latent space. By constructing cross-modal positive and negative sample pairs, this approach enhances the model's discriminative ability. Experiments on three benchmark datasets demonstrate that TriMod-DTI outperforms state-of-the-art methods. The ablation studies validate the contributions of each modality. Moreover, case studies highlight its practical potential for DTI prediction and drug discovery.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript proposes TriMod-DTI, a triple-modal contrastive learning framework for drug-target interaction (DTI) prediction. It integrates 1D sequence, 2D graph, and 3D structural features of drugs and proteins via a Feature Extractor module and a triple-modal contrastive learning strategy that aligns representations of the same entity across modalities using cross-modal positive/negative pairs. The central empirical claim is that the model outperforms state-of-the-art methods on three benchmark datasets, with ablation studies confirming the contribution of each modality and case studies illustrating practical utility for drug discovery.
Significance. If the reported gains are reproducible and arise from genuine complementarity among the three modalities rather than implementation artifacts, the work would provide a useful demonstration that 3D structural information can be productively combined with sequence and graph modalities via contrastive alignment for DTI. The ablation studies, if they include proper controls, would strengthen the case for multi-modal over uni- or bi-modal baselines. However, the absence of any quantitative metrics, dataset sizes, statistical tests, or implementation details in the provided text makes it impossible to gauge the magnitude or robustness of the claimed advance.
major comments (2)
- [Abstract] Abstract: The claim that 'Experiments on three benchmark datasets demonstrate that TriMod-DTI outperforms state-of-the-art methods' supplies no numerical results (e.g., AUC, AUPR, or accuracy deltas), no baseline names, no dataset identifiers or sizes, and no mention of statistical significance or multiple-run variance. This renders the central empirical claim unverifiable from the manuscript as presented.
- [Abstract] Abstract: The statement that 'ablation studies validate the contributions of each modality' does not report the quantitative effect sizes of removing each modality or the controls used (e.g., whether the contrastive loss is ablated independently of the feature extractor). Without these numbers, it is impossible to determine whether the modalities are complementary or largely redundant.
Simulated Author's Rebuttal
We thank the referee for these comments on the abstract. We agree that the abstract would benefit from greater specificity regarding quantitative results and controls. We will revise the abstract in the next version of the manuscript to incorporate the requested details while preserving its concise nature. Point-by-point responses follow.
read point-by-point responses
-
Referee: [Abstract] Abstract: The claim that 'Experiments on three benchmark datasets demonstrate that TriMod-DTI outperforms state-of-the-art methods' supplies no numerical results (e.g., AUC, AUPR, or accuracy deltas), no baseline names, no dataset identifiers or sizes, and no mention of statistical significance or multiple-run variance. This renders the central empirical claim unverifiable from the manuscript as presented.
Authors: We agree that the abstract, in its current form, does not include these specifics. In the revised manuscript we will update the abstract to report the key performance deltas (AUC and AUPR), the names of the primary state-of-the-art baselines, the identifiers and approximate sizes of the three benchmark datasets, and a statement on statistical significance derived from multiple independent runs with reported standard deviation. These quantitative comparisons and variance estimates already appear in the experimental results section with accompanying tables; the revision will simply surface the most salient figures in the abstract itself. revision: yes
-
Referee: [Abstract] Abstract: The statement that 'ablation studies validate the contributions of each modality' does not report the quantitative effect sizes of removing each modality or the controls used (e.g., whether the contrastive loss is ablated independently of the feature extractor). Without these numbers, it is impossible to determine whether the modalities are complementary or largely redundant.
Authors: We accept this observation. The revised abstract will include the observed effect sizes (e.g., the AUC drop when each modality is removed) and will explicitly state that the contrastive loss was ablated independently of the feature extractor to isolate its contribution. The full ablation tables, which already contain these controls and quantitative deltas, are presented in the experiments section; the abstract revision will summarize the principal effect sizes to demonstrate complementarity. revision: yes
Circularity Check
No significant circularity identified
full rationale
The paper is an empirical ML methods contribution whose central claims consist of experimental outperformance on three benchmark DTI datasets plus ablation results validating each modality. No derivation chain, equations, fitted parameters, or uniqueness theorems are present in the provided text. The performance statements are external empirical assertions that do not reduce to any self-definition, self-citation load-bearing premise, or renaming of known results; they rest on standard train/test splits and baseline comparisons outside the model's internal construction.
Axiom & Free-Parameter Ledger
Reference graph
Works this paper leans on
-
[1]
Zhao, Q., Yang, M., Cheng, Z., Li, Y., Wang, J.: Biomedical data and deep learn- ing computational models for predicting compound-protein relations. IEEE/ACM Transactions on Computational Biology and Bioinformatics19(4), 2092–2110 (2021).https://doi.org/10.1109/TCBB.2021.3069040
-
[2]
Nature Reviews Drug Discovery1(11), 882–894 (2002).https://doi.org/10.1038/nrd898
Bajorath, J.: Integration of virtual and high-throughput screening. Nature Reviews Drug Discovery1(11), 882–894 (2002).https://doi.org/10.1038/nrd898
-
[3]
In: Drug Discovery and Evaluation (2024).https://doi.org/ 10.1016/B978-0-323-99596-5.00068-8
Mak, K.-K., Wong, Y.-H., Pichika, M.R.: Artificial intelligence in drug discovery and development. In: Drug Discovery and Evaluation (2024).https://doi.org/ 10.1016/B978-0-323-99596-5.00068-8
-
[4]
Bioinformatics36(16), 4406–4414 (2020).https://doi.org/ 10.1093/bioinformatics/btaa524
Chen, L., Tan, X., Wang, D., Zhong, F., Liu, X., Yang, T., Luo, X., Chen, K., Jiang, H., Zheng, M.: TransformerCPI: improving compound–protein interaction prediction by sequence-based deep learning with self-attention mechanism and label reversal experiments. Bioinformatics36(16), 4406–4414 (2020).https://doi.org/ 10.1093/bioinformatics/btaa524
-
[5]
Bioinformatics 37(8), 1140–1147 (2021).https://doi.org/10.1093/bioinformatics/btab040
Nguyen, T., Le, H., Quinn, T.P., Nguyen, T., Le, T.D., Venkatesh, S.: GraphDTA: predicting drug–target binding affinity with graph neural networks. Bioinformatics 37(8), 1140–1147 (2021).https://doi.org/10.1093/bioinformatics/btab040
-
[6]
IEEE Transactions on Neural Networks20(1), 61–80 (2008)
Scarselli, F., Gori, M., Tsoi, A.C., Hagenbuchner, M., Monfardini, G.: The graph neural network model. IEEE Transactions on Neural Networks20(1), 61–80 (2008). https://doi.org/10.1109/TNN.2008.2005605
-
[7]
Brief- ings in Bioinformatics23(4), bbac272 (2022).https://doi.org/10.1093/bib/ bbac272
Yazdani-Jahromi, M., Yousefi, N., Tayebi, A., Kolanthai, E., Neal, C.J., Seal, S., Garibay, O.O.: AttentionSiteDTI: an interpretable graph-based model for drug- target interaction prediction using NLP sentence-level relation classification. Brief- ings in Bioinformatics23(4), bbac272 (2022).https://doi.org/10.1093/bib/ bbac272
-
[8]
Mathematical Biosciences and Engineering20(6), 10610–10625 (2023)
Wen, J., Gan, H., Yang, Z., Zhou, R., Zhao, J., Ye, Z.: Mutual-DTI: A mutual inter- action feature-based neural network for drug-target protein interaction prediction. Mathematical Biosciences and Engineering20(6), 10610–10625 (2023)
2023
-
[9]
Computational Biology and Chemistry118, 108449 (2025)
Li, C., Mi, J., Wang, H., Liu, Z., Gao, J., Wan, J.: MGMA-DTI: Drug target in- teraction prediction using multi-order gated convolution and multi-attention fusion. Computational Biology and Chemistry118, 108449 (2025)
2025
-
[10]
Semi-Supervised Classification with Graph Convolutional Networks
Kipf, T.N., Welling, M.: Semi-supervised classification with graph convolutional networks. arXiv preprint arXiv:1609.02907 (2016)
work page internal anchor Pith review Pith/arXiv arXiv 2016
-
[11]
Cheng, Z., Zhao, Q., Li, Y., Wang, J.: IIFDTI: predicting drug–target interactions through interactive and independent features based on attention mechanism. Bioin- formatics38(17), 4153–4161 (2022).https://doi.org/10.1093/bioinformatics/ btac485
-
[12]
Lin, X., Zhang, X., Yu, Z.-G., Long, Y., Zeng, X., Philip, S.Y.: CSCL-DTI: pre- dicting drug-target interaction through cross-view and self-supervised contrastive learning.In:2024IEEEInternationalConferenceonBioinformaticsandBiomedicine (BIBM), pp. 707–712. IEEE (2024).https://doi.org/10.1109/BIBM62325.2024. 10821786
-
[13]
In: International Conference on Machine Learning, pp
Radford, A., Kim, J.W., Hallacy, C., Ramesh, A., Goh, G., Agarwal, S., Sastry, G., Askell, A., Mishkin, P., Clark, J., et al.: Learning transferable visual models from natural language supervision. In: International Conference on Machine Learning, pp. 8748–8763. PMLR (2021)
2021
-
[14]
Landrum, G., et al.: RDKit: A software suite for cheminformatics, computational chemistry, and predictive modeling (2013)
2013
-
[15]
bioRxiv (2022).https://doi.org/10.1101/2022.07.21.500999
Wu, R., Ding, F., Wang, R., Shen, R., Zhang, X., Luo, S., Su, C., Wu, Z., Xie, Q., Berger, B., et al.: High-resolution de novo structure prediction from primary sequence. bioRxiv (2022).https://doi.org/10.1101/2022.07.21.500999
-
[16]
Bioinformatics37(6), 830–836 (2021)
Huang, K., Xiao, C., Glass, L.M., Sun, J.: MolTrans: molecular interaction trans- former for drug–target interaction prediction. Bioinformatics37(6), 830–836 (2021). https://doi.org/10.1093/bioinformatics/btaa880
-
[17]
In: Advances in Neural Information Processing Systems, vol
Vaswani,A.,Shazeer,N.,Parmar,N.,Uszkoreit,J.,Jones,L.,Gomez,A.N.,Kaiser, Ł., Polosukhin, I.: Attention is all you need. In: Advances in Neural Information Processing Systems, vol. 30, pp. 1–11. Curran Associates, Inc. (2017)
2017
-
[18]
arXiv preprint arXiv:2405.20358 (2024)
Mu, S., Liang, S., Li, X.: Medication Recommendation via Dual Molecular Modali- ties and Multi-Substructure Enhancement. arXiv preprint arXiv:2405.20358 (2024). https://doi.org/10.1016/j.eswa.2025.127163
-
[19]
arXiv preprint arXiv:2009.01411 (2020)
Jing, B., Eismann, S., Townshend, R.J.L., Dror, R.: Learning from protein struc- ture with geometric vector perceptrons. arXiv preprint arXiv:2009.01411 (2020). https://doi.org/10.48550/arXiv.2009.01411
-
[20]
BMC Structural Biology14, 1–9 (2014).https://doi.org/10
Saberi Fathi, S.M., Tuszynski, J.A.: A simple method for finding a protein’s ligand- binding pockets. BMC Structural Biology14, 1–9 (2014).https://doi.org/10. 1186/1472-6807-14-18
2014
-
[21]
Topology Adaptive Graph Convolutional Networks
Du, J., Zhang, S., Wu, G., Moura, J.M.F., Kar, S.: Topology adaptive graph convo- lutional networks. arXiv preprint arXiv:1710.10370 (2017).https://doi.org/10. 48550/arXiv.1710.10370
work page internal anchor Pith review Pith/arXiv arXiv 2017
-
[22]
Bioinformat- ics25(11), 1422 (2009).https://doi.org/10.1093/bioinformatics/btp163
Cock, P.J.A., Antao, T., Chang, J.T., Chapman, B.A., Cox, C.J., Dalke, A., Fried- berg, I., Hamelryck, T., Kauff, F., Wilczynski, B., et al.: Biopython: freely available Python tools for computational molecular biology and bioinformatics. Bioinformat- ics25(11), 1422 (2009).https://doi.org/10.1093/bioinformatics/btp163
-
[23]
Bioinformatics38(Supplement_2), ii68–ii74 (2022).https://doi.org/10.1093/bioinformatics/btac470
You, Y., Shen, Y.: Cross-modality and self-supervised protein embedding for com- pound–protein affinity and contact prediction. Bioinformatics38(Supplement_2), ii68–ii74 (2022).https://doi.org/10.1093/bioinformatics/btac470
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.