All-Electron Relativistic Fully Self-Consistent GW Study of Heteronuclear Actinide-Containing Diatomics
Pith reviewed 2026-06-28 19:57 UTC · model grok-4.3
The pith
Fully self-consistent GW with exact two-component relativity accurately predicts ionization energies and vibrational frequencies for uranium diatomics.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
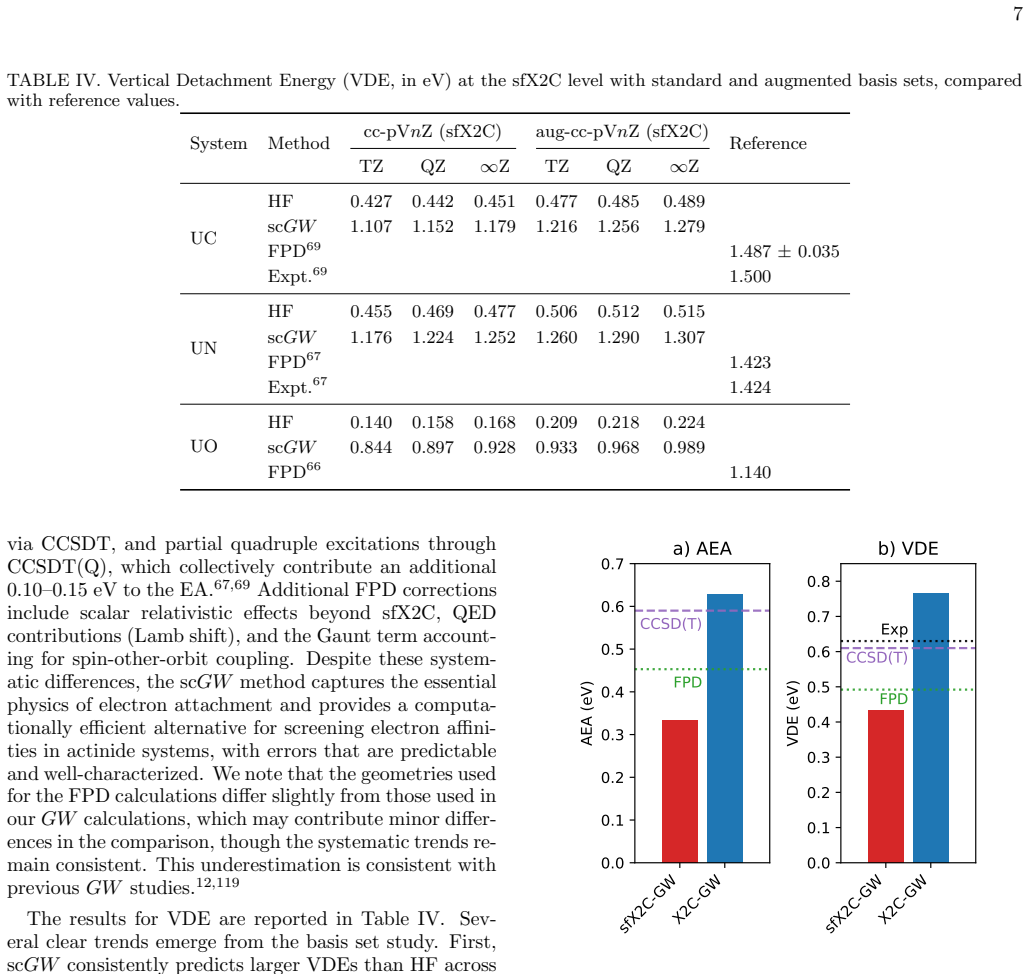
The all-electron X2C-scGW method produces ionization energies and vibrational frequencies in very good agreement with experiment and high-accuracy estimates for UC, UN, UO, and UF; electron-attachment and vertical-detachment energies require diffuse basis sets for convergence, and UF demands the two-component treatment because its attachment and detachment energies are strongly affected by spin-orbit coupling.
What carries the argument
The fully self-consistent GW approximation combined with the exact two-component (X2C) relativistic formalism, used all-electron to obtain Green's-function-based energies and spectra.
If this is right
- scGW ionization energies agree with experiment without dependence on the starting point.
- Harmonic vibrational frequencies are obtained in close agreement with benchmark values.
- Diffuse basis sets are required to converge electron-attachment and detachment energies.
- Spin-orbit coupling in UF necessitates a variational two-component relativistic treatment.
- X2C-scGW offers a practical route to actinide-molecule energetics and spectroscopy.
Where Pith is reading between the lines
- The demonstrated accuracy on diatomics supports extending the same all-electron X2C-scGW protocol to larger uranium-containing clusters or complexes.
- The strong sensitivity of UF attachment energies to spin-orbit coupling suggests that scalar-relativistic approximations will remain insufficient for many open-shell actinide systems.
- Basis-set requirements identified here indicate that future calculations on actinides must allocate resources to diffuse functions when targeting electron energetics.
Load-bearing premise
The exact two-component formalism is sufficient to capture the spin-orbit coupling effects that strongly influence electron-attachment and vertical detachment energies in UF.
What would settle it
A measured or four-component-computed electron-attachment energy for UF that deviates significantly from the X2C-scGW value would falsify the claim that the two-component treatment is adequate.
Figures
read the original abstract
The fully self-consistent $GW$ (sc$GW$) approximation provides a Green's-function approach that is starting-point independent and offers a favorable cost-to-accuracy balance compared to high-level wavefunction methods. Here, we present an all-electron sc$GW$ study of uranium-containing diatomics (UC, UN, UO, and UF), incorporating relativistic effects through the exact two-component (X2C) formalism. We evaluate adiabatic ionization energies as well as electron-attachment and detachment energetics (AEA and VDE), together with equilibrium structures and harmonic vibrational frequencies, and we assess their sensitivity to basis-set choice and relativistic treatment. We find that sc$GW$ yields ionization energies and vibrational properties in very good agreement with experiment and high-accuracy theoretical estimates. For AEA and VDE, diffuse basis sets are essential for convergence. UF is a particularly challenging case for scalar relativistic methods because its electron-attachment and vertical detachment energies are strongly affected by spin--orbit coupling, highlighting the need for a variational two-component treatment. These results establish all-electron X2C-sc$GW$ as a practical route for accurate actinide-molecule energetics and spectroscopy and motivate future applications to larger uranium-containing systems.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript applies the fully self-consistent GW (scGW) approximation in an all-electron framework with the exact two-component (X2C) relativistic treatment to the heteronuclear uranium diatomics UC, UN, UO, and UF. It computes adiabatic ionization energies, adiabatic electron affinities (AEA), vertical detachment energies (VDE), equilibrium geometries, and harmonic vibrational frequencies, while examining basis-set and relativistic-treatment sensitivities. The central claim is that scGW produces ionization energies and vibrational properties in very good agreement with experiment and high-accuracy references, that diffuse functions are required for AEA/VDE convergence, and that UF necessitates the two-component treatment because spin-orbit coupling strongly affects its electron-attachment and detachment energies.
Significance. If the numerical results bear out the stated agreement, the work is significant because it demonstrates that all-electron X2C-scGW offers a practical, starting-point-independent route to actinide-molecule energetics and spectroscopy at a cost-accuracy balance superior to high-level wavefunction methods. The explicit identification of basis-set and relativistic requirements for convergence supplies actionable guidance for the community and supports extension to larger uranium-containing systems.
Simulated Author's Rebuttal
We thank the referee for the positive assessment of our work, the recognition of its significance for actinide-molecule studies, and the recommendation to accept the manuscript.
Circularity Check
No significant circularity; direct application of established method
full rationale
The manuscript applies the pre-existing fully self-consistent GW approximation together with the exact two-component (X2C) relativistic formalism to four uranium diatomics. No new functional form, ansatz, or uniqueness theorem is derived inside the paper; all reported ionization energies, AEA/VDE values, geometries, and frequencies are numerical outputs of the standard scGW+X2C procedure run on the target molecules. Basis-set and relativistic-treatment sensitivity tests are performed against external experimental and high-level reference data rather than against quantities fitted from the same run. Consequently the central claims do not reduce to self-definition, fitted-input renaming, or load-bearing self-citation chains.
Axiom & Free-Parameter Ledger
axioms (2)
- domain assumption The fully self-consistent GW approximation is starting-point independent and provides favorable cost-to-accuracy balance.
- domain assumption The exact two-component formalism adequately incorporates relativistic effects for the studied properties.
Reference graph
Works this paper leans on
-
[1]
UC, UN and UO: Basis Set Effects UC, UN, and UO serve as a natural starting point for assessing electron attachment in uranium diatomics, be- cause the added electron primarily occupies a diffuse, largely U 7s-like orbital. Hence accurate prediction of electron affinities requires careful attention to basis set selection, as the attached electron in anion...
-
[2]
Ab-initio Green’s function meth- ods describing relativistic effects
UF: Effect of Spin-Orbit Coupling UF represents a particularly interesting test case for evaluating the treatment of spin-orbit coupling in ac- tinide systems. Upon electron attachment to form UF −, since the 7sis already doubly occupied, the additional electron occupies a uranium 6d orbital (U +[5f37s2] + e − →U[5f 36d17s2]), where strong SOC effects in ...
-
[3]
S.; Schreckenbach, G
Oakley, M. S.; Schreckenbach, G. Trends in Methanol- Solvated Actinide Ions and Actinide Expanded Por- phyrin Complexes.Inorganic Chemistry2024,64, 242– 254
-
[4]
J.; Autschbach, J.; Batista, E.; Yang, P
Duignan, T. J.; Autschbach, J.; Batista, E.; Yang, P. As- sessment of tuned range separated exchange functionals for spectroscopies and properties of uranium complexes. Journal of chemical theory and computation2017,13, 3614–3625
-
[5]
Lan, J.-H.; Wang, C.-Z.; Wu, Q.-Y.; Wang, S.-A.; Feng, Y.-X.; Zhao, Y.-L.; Chai, Z.-F.; Shi, W.-Q. A quasi-relativistic density functional theory study of the 10 actinyl (VI, V)(An= U, Np, Pu) complexes with a six- membered macrocycle containing pyrrole, pyridine, and furan subunits.The Journal of Physical Chemistry A 2015,119, 9178–9188
2015
-
[6]
Extension of the D3 and D4 London dispersion corrections to the full actinides series.Physical Chemistry Chemical Physics 2024,26, 21379–21394
Wittmann, L.; Gordiy, I.; Friede, M.; Helmich-Paris, B.; Grimme, S.; Hansen, A.; Bursch, M. Extension of the D3 and D4 London dispersion corrections to the full actinides series.Physical Chemistry Chemical Physics 2024,26, 21379–21394
2024
-
[7]
K.; Saue, T
South, C.; Shee, A.; Mukherjee, D.; Wilson, A. K.; Saue, T. 4-Component relativistic calculations of L 3 ionization and excitations for the isoelectronic species UO 2 2+, OUN+ and UN 2.Physical Chemistry Chem- ical Physics2016,18, 21010–21023
-
[8]
R.; Martin, R
Batista, E. R.; Martin, R. L.; Yang, P. Computational Studies of Bonding and Reactivity in Actinide Molec- ular Complexes.Computational Methods in Lanthanide and Actinide Chemistry2015, 375–400
-
[9]
New Method for Calculating the One-Particle Green’s Function with Application to the Electron-Gas Problem.Phys
Hedin, L. New Method for Calculating the One-Particle Green’s Function with Application to the Electron-Gas Problem.Phys. Rev.1965,139, A796–A823
1965
-
[10]
Electronic excitations: density-functional versus many-body Green’s-function approaches.Rev
Onida, G.; Reining, L.; Rubio, A. Electronic excitations: density-functional versus many-body Green’s-function approaches.Rev. Mod. Phys.2002,74, 601–659
2002
-
[11]
The GW Com- pendium: A Practical Guide to Theoretical Photoemis- sion Spectroscopy.Front
Golze, D.; Dvorak, M.; Rinke, P. The GW Com- pendium: A Practical Guide to Theoretical Photoemis- sion Spectroscopy.Front. Chem.2019,7
2019
-
[12]
Bruneval, F.; Dattani, N.; van Setten, M. J. The GW Miracle in Many-Body Perturbation Theory for the Ion- ization Potential of Molecules.Front. Chem.2021,9
2021
-
[13]
An O(N3) implementation of Hedin’s GW approximation for molecules.J
Foerster, D.; Koval, P.; S´ anchez-Portal, D. An O(N3) implementation of Hedin’s GW approximation for molecules.J. Chem. Phys.2011,135, 074105
2011
-
[14]
Resolution-of-identity approach to Hartree–Fock, hy- brid density functionals, RPA, MP2 and GW with nu- meric atom-centered orbital basis functions.New J
Ren, X.; Rinke, P.; Blum, V.; Wieferink, J.; Tkatchenko, A.; Sanfilippo, A.; Reuter, K.; Scheffler, M. Resolution-of-identity approach to Hartree–Fock, hy- brid density functionals, RPA, MP2 and GW with nu- meric atom-centered orbital basis functions.New J. Phys.2012,14, 053020
2012
-
[15]
Katharina Krause, M. E. H.; Klopper, W. Coupled- cluster reference values for the GW27 and GW100 test sets for the assessment of GW methods.Molecular Physics2015,113, 1952–1960
1952
-
[16]
J.; Rinke, P
Caruso, F.; Dauth, M.; van Setten, M. J.; Rinke, P. Benchmark of GW Approaches for the GW100 Test Set. J. Chem. Theory Comput.2016,12, 5076–5087, PMID: 27631585
2016
-
[17]
Scalable Molecular GW Calculations: Valence and Core Spectra.J
Mejia-Rodriguez, D.; Kunitsa, A.; Apr` a, E.; Govind, N. Scalable Molecular GW Calculations: Valence and Core Spectra.J. Chem. Theory Comput.2021,17, 7504– 7517, PMID: 34855381
2021
-
[18]
Robust Analytic-Continuation Approach to Many-Body GW Calculations.J
Duchemin, I.; Blase, X. Robust Analytic-Continuation Approach to Many-Body GW Calculations.J. Chem. Theory Comput.2020,16, 1742–1756, PMID: 32023052
2020
-
[19]
E.; Seiler, C.; Weigend, F.; Ev- ers, F.; van Setten, M
Kaplan, F.; Harding, M. E.; Seiler, C.; Weigend, F.; Ev- ers, F.; van Setten, M. J. Quasi-particle self-consistent GW for molecules.J. Chem. Theory Comput.2016,12, 2528–2541
2016
-
[20]
J.; Costa, R.; Vi˜ nes, F.; Illas, F
van Setten, M. J.; Costa, R.; Vi˜ nes, F.; Illas, F. Assess- ing GW Approaches for Predicting Core Level Binding Energies.J. Chem. Theory Comput.2018,14, 877–883, PMID: 29320628
2018
-
[21]
Accurate Absolute and Relative Core-Level Binding Energies from GW
Golze, D.; Keller, L.; Rinke, P. Accurate Absolute and Relative Core-Level Binding Energies from GW. J. Phys. Chem. Lett.2020,11, 1840–1847, PMID: 32043890
2020
-
[22]
S.; Conard, T.; Escudero, D.; Pourtois, G.; van Setten, M
Galleni, L.; Sajjadian, F. S.; Conard, T.; Escudero, D.; Pourtois, G.; van Setten, M. J. Modeling X-ray Pho- toelectron Spectroscopy of Macromolecules Using GW. J. Phys. Chem. Lett.2022,13, 8666–8672, PMID: 36084286
2022
-
[23]
Bench- mark of GW Methods for Core-Level Binding Energies
Li, J.; Jin, Y.; Rinke, P.; Yang, W.; Golze, D. Bench- mark of GW Methods for Core-Level Binding Energies. J. Chem. Theory Comput.2022,18, 7570–7585, PMID: 36322136
2022
-
[24]
Basis Set Selection for Molecular Core-Level GW Calcu- lations.J
Mejia-Rodriguez, D.; Kunitsa, A.; Apr` a, E.; Govind, N. Basis Set Selection for Molecular Core-Level GW Calcu- lations.J. Chem. Theory Comput.2022,18, 4919–4926, PMID: 35816679
2022
-
[25]
A.; Zgid, D.; Gull, E
Iskakov, S.; Rusakov, A. A.; Zgid, D.; Gull, E. Effect of propagator renormalization on the band gap of insulat- ing solids.Phys. Rev. B2019,100, 085112
-
[26]
Fully self- consistent finite-temperatureGWin Gaussian Bloch or- bitals for solids.Phys
Yeh, C.-N.; Iskakov, S.; Zgid, D.; Gull, E. Fully self- consistent finite-temperatureGWin Gaussian Bloch or- bitals for solids.Phys. Rev. B2022,106, 235104
-
[27]
Ab initio self- energy embedding for the photoemission spectra of NiO and MnO.Phys
Iskakov, S.; Yeh, C.-N.; Gull, E.; Zgid, D. Ab initio self- energy embedding for the photoemission spectra of NiO and MnO.Phys. Rev. B2020,102, 085105
-
[28]
N.; Shee, A.; Li, J.; Gull, E.; Zgid, D
Lan, T. N.; Shee, A.; Li, J.; Gull, E.; Zgid, D. Testing self-energy embedding theory in combination with GW. Phys. Rev. B2017,96, 155106
-
[29]
Yeh, C.-N.; Morales, M. A. Low-Scaling Algorithm for the Random Phase Approximation Using Tensor Hyper- contraction with k-point Sampling.Journal of Chemical Theory and Computation2023,19, 6197–6207
-
[30]
Yeh, C.-N.; Morales, M. A. Low-Scaling Algorithms for GW and Constrained Random Phase Approximation Using Symmetry-Adapted Interpolative Separable Den- sity Fitting.Journal of Chemical Theory and Computa- tion2024,20, 3184–3198
-
[31]
Com- pressing Green’s function using intermediate represen- tation between imaginary-time and real-frequency do- mains.Phys
Shinaoka, H.; Otsuki, J.; Ohzeki, M.; Yoshimi, K. Com- pressing Green’s function using intermediate represen- tation between imaginary-time and real-frequency do- mains.Phys. Rev. B2017,96, 035147
-
[32]
E.; van Leeuwen, R
Stan, A.; Dahlen, N. E.; van Leeuwen, R. Levels of self- consistency in the GW approximation.J. Chem. Phys. 2009,130, 114105
2009
-
[33]
W.; Thygesen, K
Rostgaard, C.; Jacobsen, K. W.; Thygesen, K. S. Fully self-consistent GW calculations for molecules.Phys. Rev. B2010,81, 085103
-
[34]
Unified description of ground and excited states of finite systems: The self-consistentGWapproach.Phys
Caruso, F.; Rinke, P.; Ren, X.; Scheffler, M.; Rubio, A. Unified description of ground and excited states of finite systems: The self-consistentGWapproach.Phys. Rev. B2012,86, 081102
-
[35]
Grumet, M.; Liu, P.; Kaltak, M.; Klimeˇ s, J. c. v.; Kresse, G. Beyond the quasiparticle approximation: Fully self-consistentGWcalculations.Phys. Rev. B 2018,98, 155143
2018
-
[36]
Strange, M.; Rostgaard, C.; H¨ akkinen, H.; Thyge- sen, K. S. Self-consistent GW calculations of electronic transport in thiol- and amine-linked molecular junc- tions.Phys. Rev. B2011,83, 115108
-
[37]
Self-consistentGW: All-electron implementa- tion with localized basis functions.Phys
Caruso, F.; Rinke, P.; Ren, X.; Rubio, A.; Schef- fler, M. Self-consistentGW: All-electron implementa- tion with localized basis functions.Phys. Rev. B2013, 88, 075105
-
[38]
Interpretation of multiple solu- 11 tions in fully iterative GF2 and GW schemes using lo- cal analysis of two-particle density matrices.J
Pokhilko, P.; Zgid, D. Interpretation of multiple solu- 11 tions in fully iterative GF2 and GW schemes using lo- cal analysis of two-particle density matrices.J. Chem. Phys.2021,155, 024101
2021
-
[39]
B.; Zgid, D
Wen, M.; Abraham, V.; Harsha, G.; Shee, A.; Wha- ley, K. B.; Zgid, D. Comparing Self-Consistent GW and Vertex-Corrected G0W0 (G0W0Γ) Accuracy for Molec- ular Ionization Potentials.J. Chem. Theory Comput. 2024,20, 3109–3120, PMID: 38573104
2024
-
[40]
A.; Zgid, D
Pokhilko, P.; Yeh, C.-N.; Morales, M. A.; Zgid, D. Ten- sor hypercontraction for fully self-consistent imaginary- time GF2 and GWSOX methods: Theory, implemen- tation, and role of the Green’s function second-order exchange for intermolecular interactions.The Journal of Chemical Physics2024,161, 084108
-
[41]
A.; Zgid, D
Pokhilko, P.; Yeh, C.-N.; Morales, M. A.; Zgid, D. Ten- sor hypercontraction for self-consistent vertex corrected GW with static and dynamic screening; applications to molecules and solids with superexchange.The Journal of Chemical Physics2025,162, 244110
-
[42]
F¨ orster, A.; Bruneval, F. Why Does the GW Approx- imation Give Accurate Quasiparticle Energies? The Cancellation of Vertex Corrections Quantified.The Journal of Physical Chemistry Letters2024,15, 12526– 12534
-
[43]
Two-Component GW Calculations: Cubic Scaling Im- plementation and Comparison of Vertex-Corrected and Partially Self-Consistent GW Variants.J
F¨ orster, A.; van Lenthe, E.; Spadetto, E.; Visscher, L. Two-Component GW Calculations: Cubic Scaling Im- plementation and Comparison of Vertex-Corrected and Partially Self-Consistent GW Variants.J. Chem. The- ory Comput.2023,19, 5958–5976, PMID: 37594901
2023
-
[44]
Beyond Quasi-Particle Self-Consistent GW for Molecules with Vertex Corrections.Journal of Chemical Theory and Computation2025,21, 1709– 1721
F¨ orster, A. Beyond Quasi-Particle Self-Consistent GW for Molecules with Vertex Corrections.Journal of Chemical Theory and Computation2025,21, 1709– 1721
-
[45]
Fully Dynamic G3W2 Self- Energy for Finite Systems: Formulas and Benchmark
Bruneval, F.; F¨ orster, A. Fully Dynamic G3W2 Self- Energy for Finite Systems: Formulas and Benchmark. Journal of Chemical Theory and Computation2024,20, 3218–3230
-
[46]
Finite temperature quantum embed- ding theories for correlated systems.New Journal of Physics2017,19, 023047
Zgid, D.; Gull, E. Finite temperature quantum embed- ding theories for correlated systems.New Journal of Physics2017,19, 023047
-
[47]
Multitier self-consistentGW+ EDMFT.Phys
Nilsson, F.; Boehnke, L.; Werner, P.; Aryasetiawan, F. Multitier self-consistentGW+ EDMFT.Phys. Rev. Mater.2017,1, 043803
2017
-
[48]
First- Principles Approach to the Electronic Structure of Strongly Correlated Systems: Combining theGWAp- proximation and Dynamical Mean-Field Theory.Phys
Biermann, S.; Aryasetiawan, F.; Georges, A. First- Principles Approach to the Electronic Structure of Strongly Correlated Systems: Combining theGWAp- proximation and Dynamical Mean-Field Theory.Phys. Rev. Lett.2003,90, 086402
2003
-
[49]
Green’s function formulation of quantum defect embedding the- ory.Journal of Chemical Theory and Computation 2022,18, 3512–3522
Sheng, N.; Vorwerk, C.; Govoni, M.; Galli, G. Green’s function formulation of quantum defect embedding the- ory.Journal of Chemical Theory and Computation 2022,18, 3512–3522
2022
-
[50]
Sakuma, R.; Friedrich, C.; Miyake, T.; Bl¨ ugel, S.; Aryasetiawan, F.GWcalculations including spin-orbit coupling: Application to Hg chalcogenides.Phys. Rev. B2011,84, 085144
-
[51]
Y.; Kotliar, G
Kutepov, A.; Haule, K.; Savrasov, S. Y.; Kotliar, G. Electronic structure of Pu and Am metals by self- consistent relativisticGWmethod.Phys. Rev. B2012, 85, 155129
-
[52]
Effect of spin-orbit interaction on the optical spectra of single-layer, double-layer, and bulk MoS2.Phys
Molina-S´ anchez, A.; Sangalli, D.; Hummer, K.; Marini, A.; Wirtz, L. Effect of spin-orbit interaction on the optical spectra of single-layer, double-layer, and bulk MoS2.Phys. Rev. B2013,88, 045412
-
[53]
GWstudy of topological insulators Bi 2Se3, Bi2Te3, and Sb2Te3: Beyond the perturbative one-shot approach
Aguilera, I.; Friedrich, C.; Bihlmayer, G.; Bl¨ ugel, S. GWstudy of topological insulators Bi 2Se3, Bi2Te3, and Sb2Te3: Beyond the perturbative one-shot approach. Phys. Rev. B2013,88, 045206
-
[54]
Relativistic GW calculations on CH3NH3PbI3 and CH3NH3SnI3 per- ovskites for solar cell applications.Sci
Umari, P.; Mosconi, E.; De Angelis, F. Relativistic GW calculations on CH3NH3PbI3 and CH3NH3SnI3 per- ovskites for solar cell applications.Sci. Rep.2014,4, 4467
2014
-
[55]
One-Electron Energies from the Two-Component GW Method.J
K¨ uhn, M.; Weigend, F. One-Electron Energies from the Two-Component GW Method.J. Chem. Theory Com- put.2015,11, 969–979, PMID: 26579751
2015
-
[56]
Imple- mentation and Validation of Fully Relativistic GW Cal- culations: Spin–Orbit Coupling in Molecules, Nanocrys- tals, and Solids.J
Scherpelz, P.; Govoni, M.; Hamada, I.; Galli, G. Imple- mentation and Validation of Fully Relativistic GW Cal- culations: Spin–Orbit Coupling in Molecules, Nanocrys- tals, and Solids.J. Chem. Theory Comput.2016,12, 3523–3544, PMID: 27331614
2016
-
[57]
Ionized, electron-attached, and excited states of molecular systems with spin–orbit cou- pling: Two-component GW and Bethe–Salpeter imple- mentations.J
Holzer, C.; Klopper, W. Ionized, electron-attached, and excited states of molecular systems with spin–orbit cou- pling: Two-component GW and Bethe–Salpeter imple- mentations.J. Chem. Phys.2019,150, 204116
2019
-
[58]
GW quasiparticle calculations with spin-orbit coupling for the light actinides.Physical Review B2014,89, 035104
Ahmed, T.; Zhu, J.-X. GW quasiparticle calculations with spin-orbit coupling for the light actinides.Physical Review B2014,89, 035104
-
[59]
I.; Rinke, P.; Schef- fler, M
Jiang, H.; Gomez-Abal, R. I.; Rinke, P.; Schef- fler, M. First-principles modeling of localized d states with the GW@ LDA+ U approach.Physical Review B—Condensed Matter and Materials Physics2010,82, 045108
-
[60]
A.; Heaven, M
Goncharov, V.; Kaledin, L. A.; Heaven, M. C. Probing the electronic structure of UO+ with high-resolution photoelectron spectroscopy.The Journal of Chemical Physics2006,125, 133202
-
[61]
Martin, J. M. L. Ab initio total atomization energies of small molecules — towards the basis set limit.Chemical Physics Letters1996,259, 669–678
-
[62]
M.; Hirao, K
Pauloviˇ c, J.; Gagliardi, L.; Dyke, J. M.; Hirao, K. A theoretical study of the gas-phase chemi-ionization re- action between uranium and oxygen atoms.The Journal of Chemical Physics2005,122, 144317
-
[63]
J.; Gibson, J
Kovacs, A.; Konings, R. J.; Gibson, J. K.; Infante, I.; Gagliardi, L. Quantum chemical calculations and ex- perimental investigations of molecular actinide oxides. Chemical Reviews2015,115, 1725–1759
-
[64]
InComprehensive Computational Chem- istry (First Edition), first edition ed.; Y´ a˜ nez, M., Boyd, R
Autschbach, J. InComprehensive Computational Chem- istry (First Edition), first edition ed.; Y´ a˜ nez, M., Boyd, R. J., Eds.; Elsevier: Oxford, 2024; pp 177–192
2024
-
[65]
Understanding covalency in molecular f-block compounds from the synergy of spectroscopy and quantum chemistry.Nature Reviews Chemistry2024,8, 701–712
Kaltsoyannis, N.; Kerridge, A. Understanding covalency in molecular f-block compounds from the synergy of spectroscopy and quantum chemistry.Nature Reviews Chemistry2024,8, 701–712
-
[66]
Dolg, M.Computational methods in lanthanide and ac- tinide chemistry; John Wiley & Sons, 2015
2015
-
[67]
Route to Chemical Accuracy for Computational Uranium Thermochemistry.Journal of Chemical Theory and Computation2022,18, 6732– 6741, Publisher: American Chemical Society
Zhang, C.; Cheng, L. Route to Chemical Accuracy for Computational Uranium Thermochemistry.Journal of Chemical Theory and Computation2022,18, 6732– 6741, Publisher: American Chemical Society
-
[68]
Romeu, J. G. F.; Hunt, A. R. E.; de Melo, G. F.; Peter- son, K. A.; Dixon, D. A. Energetic and Electronic Prop- erties of UO0/±and UF0/±.The Journal of Physical Chemistry A2024,128, 5586–5604, Publisher: Ameri- can Chemical Society
-
[69]
F.; Vasiliu, M.; Liu, G.; Ci- borowski, S.; Zhu, Z.; Blankenhorn, M.; Harris, R.; Martinez-Martinez, C.; Dipalo, M.; Peterson, K
de Melo, G. F.; Vasiliu, M.; Liu, G.; Ci- borowski, S.; Zhu, Z.; Blankenhorn, M.; Harris, R.; Martinez-Martinez, C.; Dipalo, M.; Peterson, K. A.; 12 Bowen, K. H.; Dixon, D. A. Electronic Properties of UN and UN– from Photoelectron Spectroscopy and Corre- lated Molecular Orbital Theory.The Journal of Physical Chemistry A2022,126, 7944–7953, Publisher: Amer...
-
[70]
F.; Dixon, D
de Melo, G. F.; Dixon, D. A. Bonding, Thermodynam- ics, and Spectroscopy of the Metal Borides UB0/+/– and WB0/+/–.The Journal of Physical Chemistry A 2023,127, 1588–1597, Publisher: American Chemical Society
2023
-
[71]
F.; Vasiliu, M.; Liu, G.; Ci- borowski, S.; Zhu, Z.; Blankenhorn, M.; Harris, R.; Martinez-Martinez, C.; Dipalo, M.; Peterson, K
de Melo, G. F.; Vasiliu, M.; Liu, G.; Ci- borowski, S.; Zhu, Z.; Blankenhorn, M.; Harris, R.; Martinez-Martinez, C.; Dipalo, M.; Peterson, K. A.; Bowen, K. H.; Dixon, D. A. Theoretical and Experimen- tal Study of the Spectroscopy and Thermochemistry of UC+/0/–.The Journal of Physical Chemistry A2022, 126, 9392–9407, Publisher: American Chemical Society
-
[72]
Yousfi, H.; Bensiradj, N. e. H.; Khedidji, M.; Saal, A.; Ouamerali, O. Theoretical investigation of the structure and spectroscopy of uranium oxide species.Theoretical Chemistry Accounts2023,142, 5
-
[73]
L.; Shahi, A
Infante, I.; Kovacs, A.; Macchia, G. L.; Shahi, A. R. M.; Gibson, J. K.; Gagliardi, L. Ionization energies for the actinide mono-and dioxides series, from Th to Cm: the- ory versus experiment.The Journal of Physical Chem- istry A2010,114, 6007–6015
-
[74]
O.; Heaven, M
Antonov, I. O.; Heaven, M. C. Spectroscopic and The- oretical Investigations of UF and UF+.The Journal of Physical Chemistry A2013,117, 9684–9694, Publisher: American Chemical Society
-
[75]
R.; Martin, R
Batista, E. R.; Martin, R. L.; Hay, P. J. Density func- tional investigations of the properties and thermochem- istry of UF n and UCl n (n= 1,. . . , 6).The Journal of chemical physics2004,121, 11104–11111
-
[76]
T.; Bai, X.-l.; Heaven, M
Le, A. T.; Bai, X.-l.; Heaven, M. C.; Steimle, T. C. High resolution electronic spectroscopy of uranium mononi- tride, UN.The Journal of Chemical Physics2023,158, 244301
-
[77]
J.; Morse, M
Matthew, D. J.; Morse, M. D. Resonant two-photon ion- ization spectroscopy of jet-cooled UN: Determination of the ground state.The Journal of Chemical Physics 2013,138, 184303
2013
-
[78]
R.; Bross, D
Battey, S. R.; Bross, D. H.; Peterson, K. A.; Persinger, T. D.; VanGundy, R. A.; Heaven, M. C. Spec- troscopic and theoretical studies of UN and UN+.The Journal of Chemical Physics2020,152, 094302
-
[79]
Ning, J.; Truhlar, D. G. Chemical Bonding in Isoelec- tronic NdO2 and SmO22+.The Journal of Physical Chemistry A2023,127, 2295–2305
-
[80]
Relativistic multireference quantum chemi- cal study of the electronic structure of actinide trioxide molecules.The Journal of Physical Chemistry A2017, 121, 2523–2530
Kov´ acs, A. Relativistic multireference quantum chemi- cal study of the electronic structure of actinide trioxide molecules.The Journal of Physical Chemistry A2017, 121, 2523–2530
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.