Prior-Guided Multi-Omic Transformers for Single-Cell Gene Regulatory Network Inference
Pith reviewed 2026-06-28 19:23 UTC · model grok-4.3
The pith
A Transformer learns data-driven gene-peak links from single-cell data and uses bulk-derived priors as noisy supervision to reconstruct more accurate gene regulatory networks.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
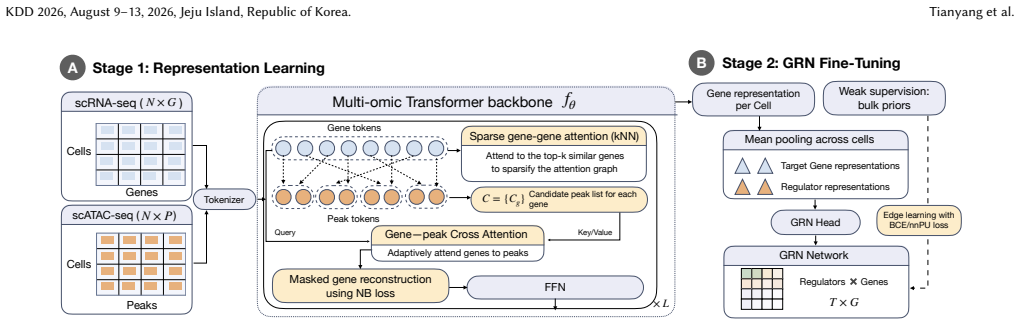
EpiAwareNet reconstructs GRNs from paired single-cell transcriptomic and chromatin accessibility data by first learning joint gene-peak representations through a gene-peak cross-attention module that performs data-driven aggregation of accessibility signals, then incorporating a bulk-derived GRN prior as noisy positive edges to provide weak supervision under label scarcity, yielding improved recovery of known regulatory interactions compared with representative baselines.
What carries the argument
Gene-peak cross-attention module for adaptive joint representations, combined with incorporation of bulk GRN prior as noisy positive edges for weak supervision.
If this is right
- EpiAwareNet improves GRN reconstruction performance over representative single- and multi-omic baselines.
- The inferred GRNs exhibit greater biological plausibility through improved recovery of known regulatory interactions.
- Lightweight biological priors from bulk data can guide single-cell GRN inference when paired with adaptive cross-modal representation learning.
- The two-stage design remains robust to noise in the supplied prior while operating under label scarcity.
Where Pith is reading between the lines
- The method could reduce reliance on large labeled single-cell datasets by leveraging existing bulk resources for other cell types or conditions.
- Similar prior-guided cross-attention designs might apply to additional multi-omic prediction tasks such as cell-type-specific chromatin modeling.
- If the noise-robustness property holds, the framework could be tested on priors derived from different tissues or species to measure transferability.
Load-bearing premise
The bulk-derived GRN prior, treated as noisy positive edges, supplies useful weak supervision that improves performance without introducing systematic bias.
What would settle it
If adding the bulk prior to the model consistently lowers recovery of held-out known interactions or increases false positives relative to the single-cell-only version, the central claim would be falsified.
Figures

read the original abstract
Gene regulatory networks (GRNs) capture transcription factor-target interactions and are central to understanding cell-state regulation and disease. Reconstructing GRNs from paired single-cell transcriptomic and chromatin accessibility data is promising but challenging: scATAC is extremely sparse, and most methods rely on fixed peak-to-gene links and weak supervision. We present EpiAwareNet, a prior-guided multi-omic Transformer framework that reconstructs GRNs from paired single-cell data using only lightweight biological priors. In Stage 1, EpiAwareNet learns joint gene-peak representations with a gene-peak cross-attention module, enabling data-driven, gene-specific aggregation of accessibility signals rather than hard-coded peak-to-gene assignments. In Stage 2, EpiAwareNet incorporates a bulk-derived GRN prior as noisy positive edges to provide weak supervision under label scarcity, refining regulatory scores while remaining robust to prior noise. In our experiments, EpiAwareNet improves GRN reconstruction over representative single- and multi-omic baselines and yields GRNs with greater biological plausibility, such as improved recovery of known regulatory interactions, suggesting that lightweight biological priors from bulk data can effectively guide single-cell GRN inference when combined with adaptive cross-modal representation learning. Code and data will be available at https://github.com/tianyang-x/EpiAwareNet_pub.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The paper presents EpiAwareNet, a two-stage prior-guided multi-omic Transformer for inferring gene regulatory networks (GRNs) from paired single-cell RNA-seq and ATAC-seq data. Stage 1 employs a gene-peak cross-attention module to learn joint representations and perform data-driven aggregation of accessibility signals, avoiding fixed peak-to-gene links. Stage 2 incorporates a bulk-derived GRN prior as noisy positive edges to supply weak supervision under label scarcity, refining regulatory scores while claiming robustness to prior noise. Experiments report improved GRN reconstruction over single- and multi-omic baselines together with greater biological plausibility via better recovery of known regulatory interactions.
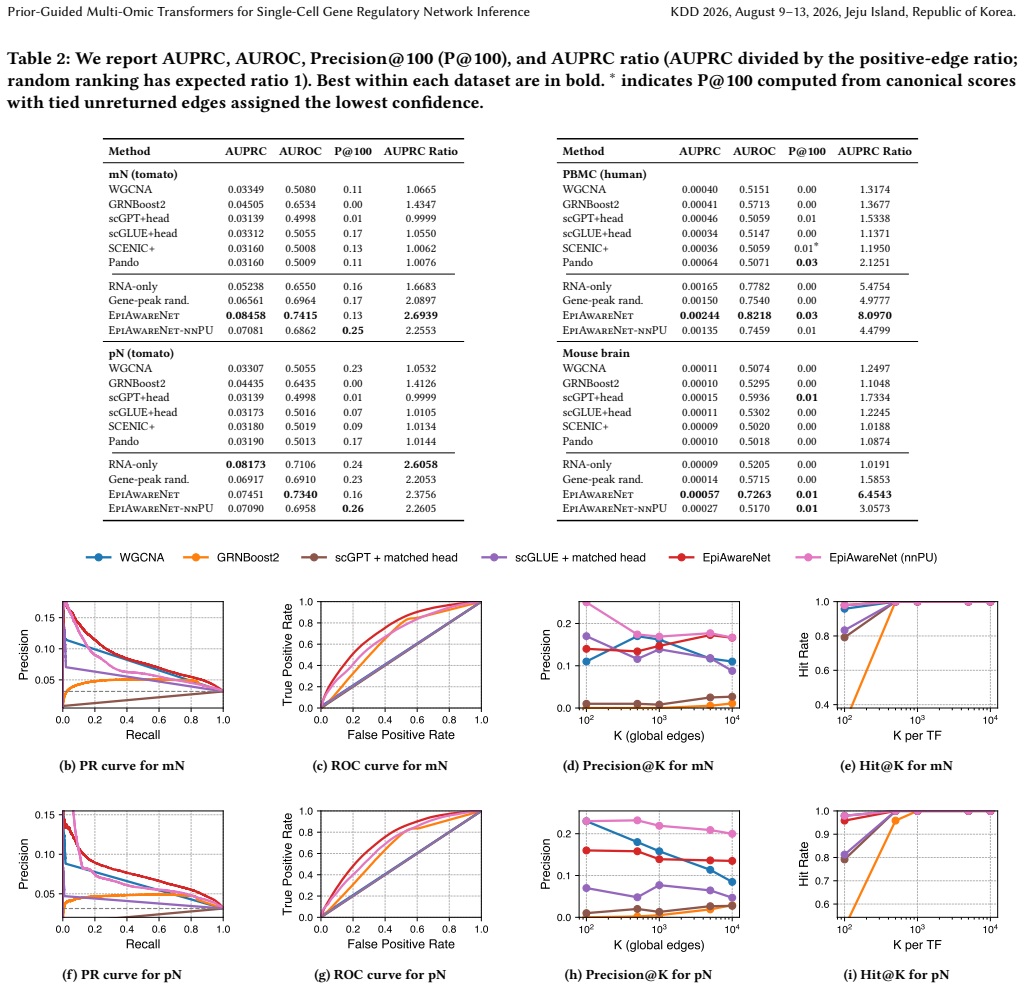
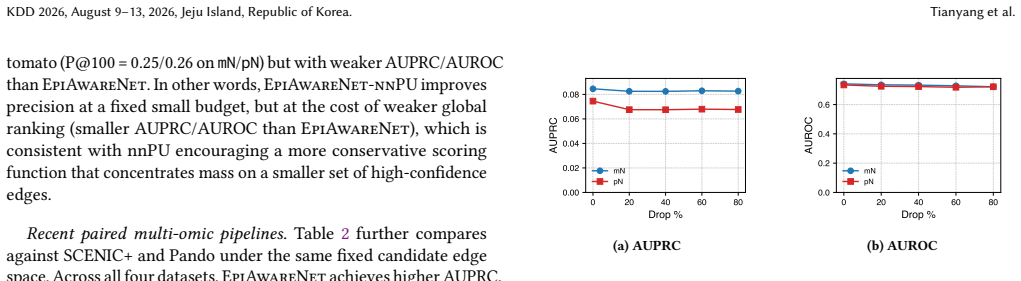
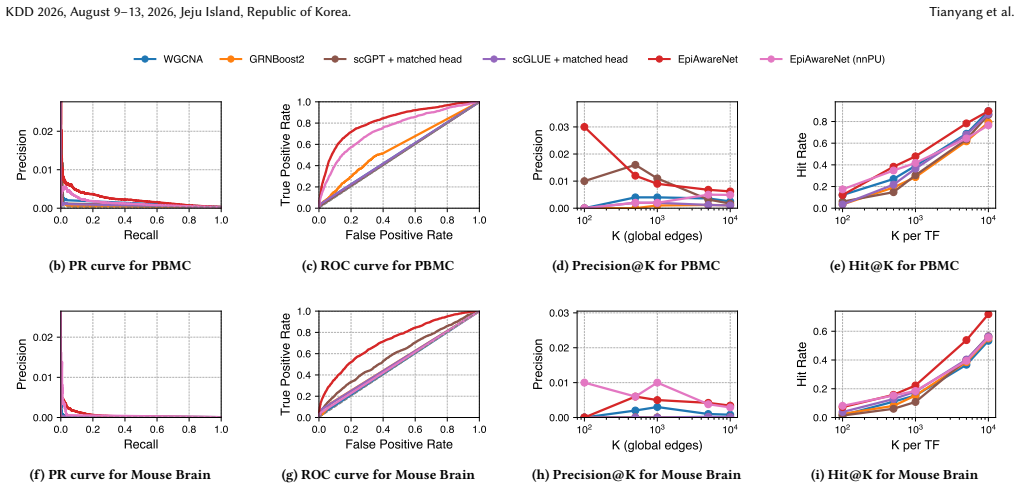
Significance. If the reported gains are shown to arise from the paired single-cell data rather than bulk prior overlap, the framework would offer a practical route to leverage lightweight external priors for single-cell GRN tasks. The adaptive cross-attention mechanism addresses a known limitation of hard-coded peak-to-gene mappings, and the emphasis on robustness to noisy priors is a useful design principle.
major comments (2)
- [Stage 2] Stage 2 (prior incorporation): the claim that the bulk GRN prior supplies useful weak supervision without introducing systematic bias toward non-cell-type-specific interactions is load-bearing for the plausibility results, yet the abstract and method description provide no quantitative ablation that isolates edges supported by the paired scRNA/scATAC data alone versus those recovered primarily through prior overlap.
- [Experiments] Experimental evaluation: the reported improvements in GRN reconstruction and recovery of known interactions lack visible details on data splits, multiple-testing correction, or statistical tests that would confirm the gains survive correction; without these, it is unclear whether the central claim of outperformance is robust.
minor comments (1)
- The abstract states that code and data will be available at a GitHub link; confirming that the repository contains the exact data splits and evaluation scripts used in the reported experiments would strengthen reproducibility.
Simulated Author's Rebuttal
We thank the referee for the detailed and constructive review. We address each major comment below, clarifying our approach and outlining planned revisions to strengthen the manuscript.
read point-by-point responses
-
Referee: [Stage 2] Stage 2 (prior incorporation): the claim that the bulk GRN prior supplies useful weak supervision without introducing systematic bias toward non-cell-type-specific interactions is load-bearing for the plausibility results, yet the abstract and method description provide no quantitative ablation that isolates edges supported by the paired scRNA/scATAC data alone versus those recovered primarily through prior overlap.
Authors: We agree that an explicit ablation isolating the contribution of the paired single-cell data from the bulk prior is important to substantiate the claim of robustness and to rule out systematic bias from prior overlap. In the revised manuscript we will add a new ablation experiment comparing (i) the full EpiAwareNet model, (ii) a variant trained without the prior (using only the cross-attention representations from paired scRNA/scATAC), and (iii) a prior-only baseline. We will report the fraction of recovered edges unique to the single-cell data, the overlap with the bulk prior, and performance on cell-type-specific benchmarks to quantify the added value of the paired data. revision: yes
-
Referee: [Experiments] Experimental evaluation: the reported improvements in GRN reconstruction and recovery of known interactions lack visible details on data splits, multiple-testing correction, or statistical tests that would confirm the gains survive correction; without these, it is unclear whether the central claim of outperformance is robust.
Authors: We acknowledge that the current manuscript does not provide sufficient detail on the evaluation protocol. In the revision we will expand the Experiments section to include: (1) explicit description of data splits (e.g., cell-wise or gene-wise train/validation/test partitions and how they avoid leakage), (2) the multiple-testing correction procedure applied to reported metrics, and (3) the statistical tests (with p-values) used to compare EpiAwareNet against baselines. These additions will allow readers to assess the robustness of the reported gains. revision: yes
Circularity Check
No circularity: empirical ML framework with external validation
full rationale
The paper describes a two-stage Transformer model for GRN inference that learns representations via cross-attention in Stage 1 and applies a bulk-derived prior as weak supervision in Stage 2. No equations, derivations, or fitted parameters are presented that reduce any reported improvement or plausibility metric to a quantity defined by the prior itself. Claims rest on comparisons to external baselines and recovery of known interactions, with no self-citation load-bearing steps or self-definitional constructions visible in the provided text. The approach is self-contained against independent benchmarks.
Axiom & Free-Parameter Ledger
Reference graph
Works this paper leans on
-
[1]
Kyle Akers and T. M. Murali. 2021. Gene regulatory network inference in single- cell biology.Current Opinion in Systems Biology26 (2021), 87–97. doi:10.1016/j. coisb.2021.04.007
work page doi:10.1016/j 2021
-
[2]
Carmen Bravo González-Blas et al. 2023. SCENIC+: single-cell multiomic infer- ence of enhancers and gene regulatory networks.Nature Methods20, 9 (2023), 1355–1367. doi:10.1038/s41592-023-01938-4
-
[3]
Zhi-Jie Cao and Ge Gao. 2022. Multi-omics single-cell data integration and regulatory inference with graph-linked embedding.Nature Biotechnology40, 10 (2022), 1458–1466. doi:10.1038/s41587-022-01284-4
-
[4]
Shuonan Chen and Jessica C. Mar. 2018. Evaluating methods of inferring gene regulatory networks highlights their lack of performance for single cell gene 9 KDD 2026, August 9–13, 2026, Jeju Island, Republic of Korea. Tianyang et al. expression data.BMC Bioinformatics19, 1 (2018), 232. doi:10.1186/s12859-018- 2217-z
-
[5]
Haotian Cui, Chloe Wang, Hassaan Maan, Kuan Pang, Fengning Luo, Nan Duan, and Bo Wang. 2024. scGPT: toward building a foundation model for single- cell multi-omics using generative AI.Nature Methods21, 8 (2024), 1470–1480. doi:10.1038/s41592-024-02201-0
- [6]
-
[7]
Riet De Smet and Kathleen Marchal. 2010. Advantages and limitations of current network inference methods.Nature Reviews Microbiology8, 10 (2010), 717–729. doi:10.1038/nrmicro2419
-
[8]
Jeremiah J. Faith, Boris Hayete, Joshua T. Thaden, Ilaria Mogno, Jeffrey Wierzbowski, Gilles Cottarel, Simon Kasif, James J. Collins, and Timothy S. Gard- ner. 2007. Large-Scale Mapping and Validation ofEscherichia coliTranscriptional Regulation from a Compendium of Expression Profiles.PLoS Biology5, 1 (2007), e8. doi:10.1371/journal.pbio.0050008
-
[9]
Gray Camp, and Barbara Treutlein
Jonas Simon Fleck, Sophie Martina Johanna Jansen, Damian Wollny, Fides Zenk, Makiko Seimiya, Akanksha Jain, Ryoko Okamoto, Malgorzata Santel, Zhisong He, J. Gray Camp, and Barbara Treutlein. 2023. Inferring and perturbing cell fate regulomes in human brain organoids.Nature621, 7978 (2023), 365–372. doi:10.1038/s41586-022-05279-8
-
[10]
Luz Garcia-Alonso, Christian H. Holland, Mahmoud M. Ibrahim, Denes Turei, and Julio Saez-Rodriguez. 2019. Benchmark and integration of resources for the estimation of human transcription factor activities.Genome Research29, 8 (2019), 1363–1375. doi:10.1101/gr.240663.118
-
[11]
Heonjong Han, Jae-Won Cho, Sangyoung Lee, Ayoung Yun, Hyojin Kim, Dasom Bae, Sunmo Yang, Chan Yeong Kim, Muyoung Lee, Eunbeen Lee, Sungho Lee, Byunghee Kang, Dabin Jeong, Yaeji Kim, Hyeon-Nae Jeon, Haein Jung, Sunhwee Nam, Michael Chung, Jong-Hoon Kim, and Insuk Lee. 2018. TRRUST v2: an expanded reference database of human and mouse transcriptional regula...
-
[12]
Mauck III, Shiwei Zheng, Andrew Butler, Maddie J
Yuhan Hao, Stephanie Hao, Erica Andersen-Nissen, William M. Mauck III, Shi- wei Zheng, Andrew Butler, Maddie J. Lee, Aleksander J. Wilk, Charlotte Darby, Michael Zager, Paul Hoffman, Marlon Stoeckius, Efthymia Papalexi, Eleni P. Mim- itou, Jay A. Jain, Avi Srivastava, et al. 2021. Integrated analysis of multimodal single-cell data.Cell184, 13 (2021), 3573...
-
[13]
Vân Anh Huynh-Thu, Alexandre Irrthum, Louis Wehenkel, and Pierre Geurts
-
[14]
doi:10.1371/journal.pone.0012776
Inferring regulatory networks from expression data using tree-based methods.PLoS ONE5, 9 (2010), e12776. doi:10.1371/journal.pone.0012776
-
[15]
Guy Karlebach and Ron Shamir. 2008. Modelling and analysis of gene regulatory networks.Nature Reviews Molecular Cell Biology9, 10 (2008), 770–780. doi:10. 1038/nrm2503
2008
-
[16]
Kharchenko, Lev Silberstein, and David T
Peter V. Kharchenko, Lev Silberstein, and David T. Scadden. 2014. Bayesian approach to single-cell differential expression analysis.Nature Methods11, 7 (2014), 740–742. doi:10.1038/nmeth.2967
-
[17]
du Plessis, and Masashi Sugiyama
Ryuichi Kiryo, Gang Niu, Marthinus C. du Plessis, and Masashi Sugiyama. 2017. Positive-unlabeled learning with non-negative risk estimator. InAdvances in Neural Information Processing Systems, Vol. 30. Curran Associates, Inc., Red Hook, NY, USA, 1675–1685
2017
-
[19]
Evan Z. Macosko, Anindita Basu, Rahul Satija, James Nemesh, Karthik Shekhar, Melissa Goldman, Itay Tirosh, Allison R. Bialas, Nolan Kamitaki, Emily M. Marter- steck, John J. Trombetta, David A. Weitz, Joshua R. Sanes, Alex K. Shalek, Aviv Regev, and Steven A. McCarroll. 2015. Highly Parallel Genome-wide Expres- sion Profiling of Individual Cells Using Nan...
-
[20]
Adam A. Margolin, Ilya Nemenman, Katia Basso, Chris Wiggins, Gustavo Stolovitzky, Riccardo Dalla Favera, and Andrea Califano. 2006. ARACNE: An Algorithm for the Reconstruction of Gene Regulatory Networks in a Mammalian Cellular Context.BMC Bioinformatics7, Suppl 1 (2006), S7. doi:10.1186/1471- 2105-7-S1-S7
-
[21]
Thomas Moerman, Sara Aibar, Carmen Bravo González-Blas, Jaak Simm, Yves Moreau, Jan Aerts, and Stein Aerts. 2019. GRNBoost2 and Arboreto: efficient and scalable inference of gene regulatory networks.Bioinformatics35, 12 (2019), 2159–2161. doi:10.1093/bioinformatics/bty916
-
[22]
Ryan M Patrick, Rajeev Ranjan, Shantha R Sumanasinghe, Phillip M San Miguel, Kranthi Varala, and Ying Li. 2026. Every Cell Counts: Tomato Root Responses to Nitrogen at Single-Cell Resolution.bioRxiv(2026). arXiv:https://www.biorxiv.org/content/early/2026/02/07/2026.02.06.704465.full.pdf doi:10.64898/2026.02.06.704465
-
[23]
Griffiths, Caroline Guibentif, Thomas W
Blanca Pijuan-Sala, Jonathan A. Griffiths, Caroline Guibentif, Thomas W. Hiscock, Wardha Jawaid, Fernando J. Calero-Nieto, Carla Mulas, Ximena Ibarra-Soria, Richard C. V. Tyser, Dominic L. L. Ho, Wolf Reik, Shankar Srinivas, Benjamin D. Simons, Jennifer Nichols, John C. Marioni, and Berthold Gottgens. 2019. A single- cell molecular map of mouse gastrulati...
-
[24]
Samantha A. Pliner, Jonathan S. Packer, José L. McFaline-Figueroa, Darren A. Cusanovich, Riza M. Daza, Ilya Aigha, Charles P. Fulco, Jason H. Bielas, Sebastian Preissl, Grace X. Y. Zheng, and Cole Trapnell. 2018. Cicero predicts cis-regulatory DNA interactions from single-cell chromatin accessibility data.Molecular Cell 71, 5 (2018), 858–871.e8. doi:10.10...
-
[25]
Jalihal, Jeffrey N
Aditya Pratapa, Amogh P. Jalihal, Jeffrey N. Law, Aditya Bharadwaj, and T. M. Murali. 2020. Benchmarking algorithms for gene regulatory network inference from single-cell transcriptomic data.Nature Methods17, 2 (2020), 147–154. doi:10. 1038/s41592-019-0690-6
2020
-
[26]
Anthony N. Schep, Botao Wu, Jason D. Buenrostro, and William J. Greenleaf. 2017. chromVAR: inferring transcription-factor-associated accessibility from single-cell epigenomic data.Nature Methods14, 10 (2017), 975–978. doi:10.1038/nmeth.4401
-
[27]
Stefan Schoenfelder and Peter Fraser. 2019. Long-range enhancer–promoter contacts in gene expression control.Nature Reviews Genetics20, 8 (2019), 437–
2019
-
[28]
doi:10.1038/s41576-019-0128-0
-
[29]
Marco Stock, Corinna Losert, Matteo Zambon, Niclas Popp, Gabriele Lubatti, Eva Hörmanseder, Matthias Heinig, and Antonio Scialdone. 2025. Leveraging prior knowledge to infer gene regulatory networks from single-cell RNA-sequencing data.Molecular Systems Biology21, 3 (2025), 214–230. doi:10.1038/s44320-025- 00088-3
-
[30]
Mauck, Yuhan Hao, Marlon Stoeckius, Peter Smibert, and Rahul Satija
Tim Stuart, Andrew Butler, Paul Hoffman, Christoph Hafemeister, Efthymia Papalexi, William M. Mauck III, Yuhan Hao, Marlon Stoeckius, Peter Smibert, and Rahul Satija. 2019. Comprehensive integration of single-cell data.Cell177, 7 (2019), 1888–1902.e21. doi:10.1016/j.cell.2019.05.031
-
[31]
Theodoris, Ling Xiao, Anant Chopra, Mark D
Christina V. Theodoris, Ling Xiao, Anant Chopra, Mark D. Chaffin, Zeina R. Al Sayed, Matthew C. Hill, Helene Mantineo, Elizabeth M. Brydon, Zexian Zeng, X. Shirley Liu, and Patrick T. Ellinor. 2023. Transfer learning enables predictions in network biology.Nature618, 7965 (2023), 616–624. doi:10.1038/s41586-023- 06139-9
-
[32]
Cole Trapnell, Davide Cacchiarelli, Justin Grimsby, Pallavi Pokharel, Shuqiang Li, Michael Morse, Niall J. Lennon, Kenneth J. Livak, Tarjei S. Mikkelsen, and John L. Rinn. 2014. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells.Nature Biotechnology32 (2014), 381–386. doi:10.1038/nbt.2859
-
[33]
Grace X. Y. Zheng, Julie M. Terry, Phillip Belgrader, Paul Ryvkin, Zachary W. Bent, Robert Wilson, Sean B. Ziraldo, Tobias D. Wheeler, Geoff P. McDermott, Junjie Zhu, Michael T. Gregory, Joe Shuga, Lydia Montesclaros, James G. Under- wood, Donald A. Masquelier, Sara Y. Nishimura, Michael Schnall-Levin, Peter W. Wyatt, Colin M. Hindson, Rajiv Bharadwaj, Al...
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.