CryoProt: A Protein Pretraining Framework with Cross-Box Interactions on Cryo-EM Density Maps
Pith reviewed 2026-06-28 17:49 UTC · model grok-4.3
The pith
CryoProt pretrains protein representations from cryo-EM density maps by letting local boxes interact through a shared latent space.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
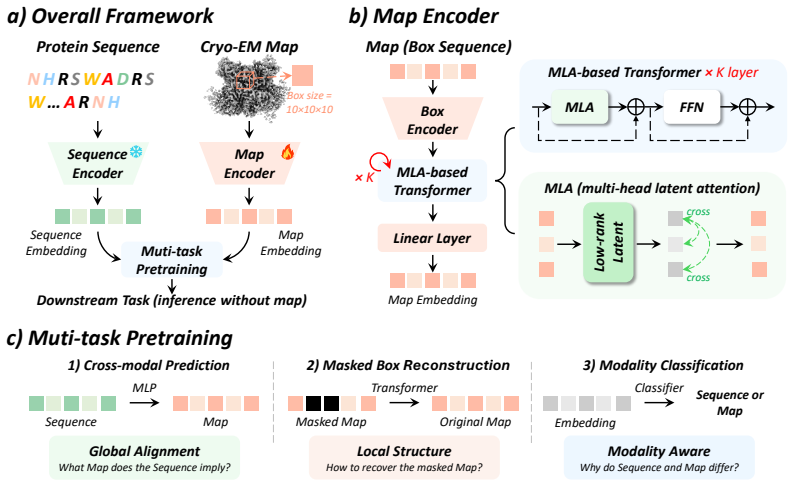
CryoProt's Map Encoder applies multi-head latent attention so that box-level representations interact via a shared latent space, explicitly modeling cross-box dependencies within the density map, and combines this with multi-task pretraining to produce representations that transfer to diverse protein tasks without requiring density maps at inference.
What carries the argument
Map Encoder based on multi-head latent attention, which routes box-level representations through a shared latent space to capture cross-box dependencies.
If this is right
- Representations learned during pretraining transfer directly to protein flexibility prediction and similar tasks without density maps at test time.
- Explicit modeling of cross-box interactions improves performance over methods that treat boxes independently.
- Multi-task pretraining on cryo-EM maps produces generalizable features usable across multiple protein property prediction problems.
- Gains of up to 12 percent over prior state-of-the-art baselines are observed on the reported benchmarks.
Where Pith is reading between the lines
- The same latent-space interaction pattern could be tested on other forms of 3D structural imaging data.
- Pretraining that implicitly encodes global context may lower the labeled data needed for related protein tasks.
- Hybrid models that combine this encoder with sequence-only pretraining could be evaluated for further gains.
Load-bearing premise
The multi-head latent attention mechanism captures the essential cross-box dependencies that improve representation quality for transfer to tasks that do not supply density maps at inference.
What would settle it
A version of the model that removes the cross-box interaction component of the Map Encoder and still matches or exceeds CryoProt's benchmark scores would falsify the central claim.
Figures
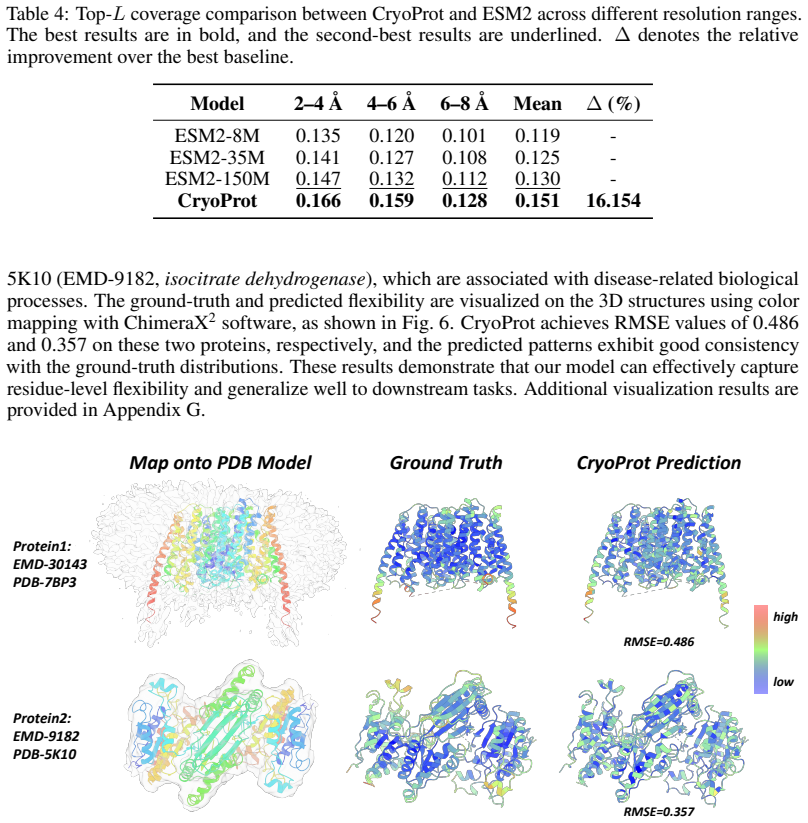
read the original abstract
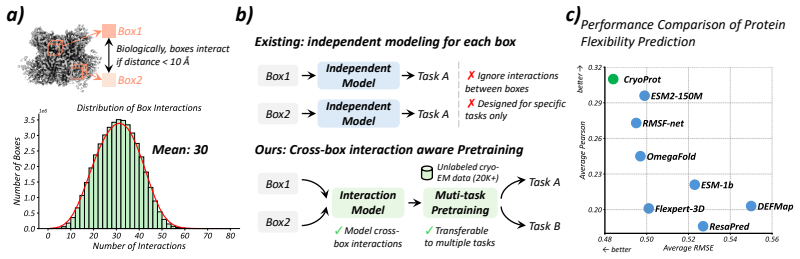
Despite the growing availability of cryo-electron microscopy (cryo-EM) density maps, effectively leveraging them for protein representation remains challenging. First, current methods lack a general-purpose protein pretraining framework tailored for cryo-EM density maps, designed for protein-related property prediction. Second, existing approaches typically partition density maps into local box regions and model them independently, overlooking interactions across boxes which are essential for capturing global structural context in cryo-EM density map. To address these challenges, we propose CryoProt, a protein pretraining framework designed for cryo-EM density maps. CryoProt introduces a Map Encoder based on multi-head latent attention (MLA), where box-level representations interact through a shared latent space, enabling explicit modeling of cross-box dependencies within the density map. Furthermore, we adopt a multi-task pretraining strategy to learn generalizable representations that can be effectively transferred to diverse downstream tasks, such as protein flexibility prediction, where cryo-EM density maps are not required and can be inferred implicitly by the pretrained model. Experimental results demonstrate that CryoProt consistently outperforms existing state-of-the-art methods across multiple benchmarks, achieving up to 12% improvement over the best-performing baselines, highlighting the importance of modeling cross-box interactions in cryo-EM data. The source code is publicly available at https://anonymous.4open.science/r/CryoProt.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript proposes CryoProt, a pretraining framework for protein representations from cryo-EM density maps. It introduces a Map Encoder using multi-head latent attention (MLA) to allow box-level features to interact via a shared latent space, thereby modeling cross-box dependencies that prior independent-box approaches overlook. A multi-task pretraining objective produces transferable representations usable on downstream tasks (e.g., protein flexibility prediction) without requiring density maps at inference time. Experiments report consistent gains over existing methods, reaching up to 12% improvement, and the source code is released publicly.
Significance. If the performance attribution holds, the framework would supply a general-purpose pretraining recipe that explicitly incorporates global structural context from cryo-EM maps while remaining applicable to tasks lacking map input. Public code availability is a clear strength that aids reproducibility and follow-up work.
major comments (1)
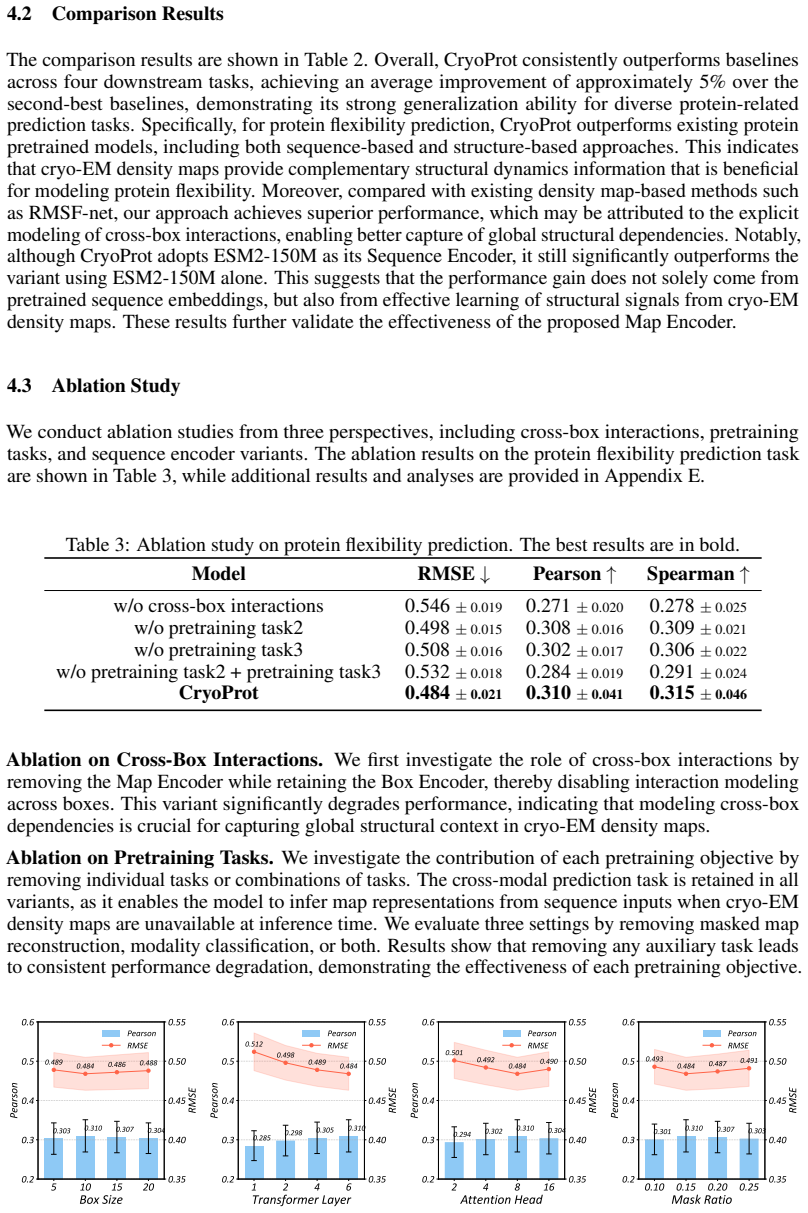
- [Experimental evaluation section] Experimental evaluation section: no ablation is presented that isolates the MLA cross-box interaction (e.g., by replacing the latent attention with independent per-box processing while freezing pretraining tasks, data, and all other architectural choices). Without this controlled comparison, the reported 12% gains cannot be confidently attributed to cross-box modeling rather than multi-task pretraining or other factors, undermining the central claim.
Simulated Author's Rebuttal
We thank the referee for the detailed and constructive review. The central concern regarding the lack of a controlled ablation isolating the multi-head latent attention (MLA) component is valid and directly addresses the attribution of performance gains. We address this point below and commit to incorporating the requested experiment.
read point-by-point responses
-
Referee: [Experimental evaluation section] Experimental evaluation section: no ablation is presented that isolates the MLA cross-box interaction (e.g., by replacing the latent attention with independent per-box processing while freezing pretraining tasks, data, and all other architectural choices). Without this controlled comparison, the reported 12% gains cannot be confidently attributed to cross-box modeling rather than multi-task pretraining or other factors, undermining the central claim.
Authors: We agree that a direct ablation isolating the cross-box interaction mechanism is necessary to strengthen the causal attribution. In the revised manuscript we will add a controlled ablation that replaces the MLA module with independent per-box processing (i.e., no latent-space interaction) while keeping the pretraining tasks, training data, optimizer, and all other architectural hyperparameters identical. This will allow quantitative measurement of the incremental benefit attributable to cross-box modeling. We will report the resulting performance delta on the same downstream benchmarks used in the original experiments. revision: yes
Circularity Check
No circularity: empirical framework with independent design choices
full rationale
The paper introduces CryoProt as a new pretraining framework using a Map Encoder with multi-head latent attention for cross-box interactions plus multi-task learning, evaluated on downstream benchmarks. No derivation chain, mathematical prediction, or first-principles result is presented that reduces to its own inputs by construction. The abstract and described claims contain no self-citations, no fitted parameters renamed as predictions, and no uniqueness theorems imported from prior author work. Performance improvements are asserted via experimental comparison rather than logical equivalence to the input data or architecture. This is a standard empirical ML contribution whose validity rests on external benchmarks, not internal definitional closure.
Axiom & Free-Parameter Ledger
Reference graph
Works this paper leans on
-
[1]
Poet: A generative model of protein families as sequences-of-sequences
Timothy Truong Jr and Tristan Bepler. Poet: A generative model of protein families as sequences-of-sequences. InAdvances in Neural Information Processing Systems, volume 36, pages 77379–77415, 2023
2023
-
[2]
The topological properties of the protein universe
Christian D Madsen, Agnese Barbensi, Stephen Y Zhang, Lucy Ham, Alessia David, Dou- glas EV Pires, and Michael PH Stumpf. The topological properties of the protein universe. Nature Communications, 16(1):7503, 2025
2025
-
[3]
Boosting the predictive power of protein representations with a corpus of text annotations.Nature Machine Intelligence, 7(9):1403–1413, 2025
Haonan Duan, Marta Skreta, Leonardo Cotta, Ella Miray Rajaonson, Nikita Dhawan, Alán Aspuru-Guzik, and Chris J Maddison. Boosting the predictive power of protein representations with a corpus of text annotations.Nature Machine Intelligence, 7(9):1403–1413, 2025
2025
-
[4]
Learning meaningful represen- tations of protein sequences.Nature communications, 13(1):1914, 2022
Nicki Skafte Detlefsen, Søren Hauberg, and Wouter Boomsma. Learning meaningful represen- tations of protein sequences.Nature communications, 13(1):1914, 2022
1914
-
[5]
Copra: Bridging cross-domain pretrained sequence models with complex structures for protein-rna binding affinity prediction
Rong Han, Xiaohong Liu, Tong Pan, Jing Xu, Xiaoyu Wang, Wuyang Lan, Zhenyu Li, Zixuan Wang, Jiangning Song, Guangyu Wang, et al. Copra: Bridging cross-domain pretrained sequence models with complex structures for protein-rna binding affinity prediction. InProceedings of the AAAI Conference on Artificial Intelligence, volume 39, pages 246–254, 2025
2025
-
[6]
Msa transformer
Roshan M Rao, Jason Liu, Robert Verkuil, Joshua Meier, John Canny, Pieter Abbeel, Tom Sercu, and Alexander Rives. Msa transformer. InInternational Conference on Machine Learning, pages 8844–8856. PMLR, 2021
2021
-
[7]
Protein structure tok- enization: Benchmarking and new recipe
Xinyu Yuan, Zichen Wang, Marcus D Collins, and Huzefa Rangwala. Protein structure tok- enization: Benchmarking and new recipe. InInternational Conference on Machine Learning, pages 73645–73670. PMLR, 2025
2025
-
[8]
Data-driven regularization lowers the size barrier of cryo-em structure determination.Nature Methods, 21(7):1216–1221, 2024
Dari Kimanius, Kiarash Jamali, Max E Wilkinson, Sofia Lövestam, Vaithish Velazhahan, Takanori Nakane, and Sjors HW Scheres. Data-driven regularization lowers the size barrier of cryo-em structure determination.Nature Methods, 21(7):1216–1221, 2024
2024
-
[9]
Accurate global and local 3d alignment of cryo-em density maps using local spatial structural features.Nature Communications, 15(1):1593, 2024
Bintao He, Fa Zhang, Chenjie Feng, Jianyi Yang, Xin Gao, and Renmin Han. Accurate global and local 3d alignment of cryo-em density maps using local spatial structural features.Nature Communications, 15(1):1593, 2024
2024
-
[10]
arXiv preprint arXiv:2506.04490 , year=
Rishwanth Raghu, Axel Levy, Gordon Wetzstein, and Ellen D Zhong. Multiscale guidance of protein structure prediction with heterogeneous cryo-em data.arXiv preprint arXiv:2506.04490, 2025
-
[11]
Deepemhancer: a deep learning solution for cryo-em volume post-processing.Communications biology, 4(1):874, 2021
Ruben Sanchez-Garcia, Josue Gomez-Blanco, Ana Cuervo, Jose Maria Carazo, Carlos Oscar S Sorzano, and Javier Vargas. Deepemhancer: a deep learning solution for cryo-em volume post-processing.Communications biology, 4(1):874, 2021
2021
-
[12]
Cryoalign2: efficient global and local cryo-em map retrieval based on parallel-accelerated local spatial structural features.Bioinformatics, 41(5):btaf296, 2025
Zhe Liu, Bintao He, Tian Zhang, Chenjie Feng, Fa Zhang, Zhongjun Yang, and Renmin Han. Cryoalign2: efficient global and local cryo-em map retrieval based on parallel-accelerated local spatial structural features.Bioinformatics, 41(5):btaf296, 2025. 10
2025
-
[13]
Extraction of protein dynamics information from cryo-em maps using deep learning.Nature Machine Intelligence, 3(2):153–160, 2021
Shigeyuki Matsumoto, Shoichi Ishida, Mitsugu Araki, Takayuki Kato, Kei Terayama, and Yasushi Okuno. Extraction of protein dynamics information from cryo-em maps using deep learning.Nature Machine Intelligence, 3(2):153–160, 2021
2021
-
[14]
Xintao Song, Lei Bao, Chenjie Feng, Qiang Huang, Fa Zhang, Xin Gao, and Renmin Han. Accurate prediction of protein structural flexibility by deep learning integrating intricate atomic structures and cryo-em density information.Nature Communications, 15(1):5538, 2024
2024
-
[15]
Atlas: protein flexibility description from atomistic molecular dynamics simulations
Yann Vander Meersche, Gabriel Cretin, Aria Gheeraert, Jean-Christophe Gelly, and Tatiana Ga- lochkina. Atlas: protein flexibility description from atomistic molecular dynamics simulations. Nucleic acids research, 52(D1):D384–D392, 2024
2024
-
[16]
Protein complex structure modeling by cross-modal alignment between cryo-em maps and protein sequences.Nature Communications, 15(1):8808, 2024
Sheng Chen, Sen Zhang, Xiaoyu Fang, Liang Lin, Huiying Zhao, and Yuedong Yang. Protein complex structure modeling by cross-modal alignment between cryo-em maps and protein sequences.Nature Communications, 15(1):8808, 2024
2024
-
[17]
Cryoten: efficiently enhancing cryo-em density maps using transformers.Bioinformatics, 41(3):btaf092, 2025
Joel Selvaraj, Liguo Wang, and Jianlin Cheng. Cryoten: efficiently enhancing cryo-em density maps using transformers.Bioinformatics, 41(3):btaf092, 2025
2025
-
[18]
Cryofm: A flow-based foundation model for cryo-em densities.arXiv preprint arXiv:2410.08631, 2024
Yi Zhou, Yilai Li, Jing Yuan, and Quanquan Gu. Cryofm: A flow-based foundation model for cryo-em densities.arXiv preprint arXiv:2410.08631, 2024
-
[19]
Gil Koren, Sagi Meir, Lennard Holschuh, Haydyn DT Mertens, Tamara Ehm, Nadav Yahalom, Adina Golombek, Tal Schwartz, Dmitri I Svergun, Omar A Saleh, et al. Intramolecular structural heterogeneity altered by long-range contacts in an intrinsically disordered protein.Proceedings of the National Academy of Sciences, 120(30):e2220180120, 2023
2023
-
[20]
Highly accurate protein structure prediction with alphafold.Nature, 596(7873):583–589, 2021
John Jumper, Richard Evans, Alexander Pritzel, Tim Green, Michael Figurnov, Olaf Ron- neberger, Kathryn Tunyasuvunakool, Russ Bates, Augustin Žídek, Anna Potapenko, et al. Highly accurate protein structure prediction with alphafold.Nature, 596(7873):583–589, 2021
2021
-
[21]
Alexander Rives, Joshua Meier, Tom Sercu, Siddharth Goyal, Zeming Lin, Jason Liu, Demi Guo, Myle Ott, C Lawrence Zitnick, Jerry Ma, et al. Biological structure and function emerge from scaling unsupervised learning to 250 million protein sequences.Proceedings of the National Academy of Sciences, 118(15):e2016239118, 2021
2021
-
[22]
Language models enable zero-shot prediction of the effects of mutations on protein function
Joshua Meier, Roshan Rao, Robert Verkuil, Jason Liu, Tom Sercu, and Alex Rives. Language models enable zero-shot prediction of the effects of mutations on protein function. InAdvances in Neural Information Processing Systems, volume 34, pages 29287–29303, 2021
2021
-
[23]
Language models of protein sequences at the scale of evolution enable accurate structure prediction.BioRxiv, 2022: 500902, 2022
Zeming Lin, Halil Akin, Roshan Rao, Brian Hie, Zhongkai Zhu, Wenting Lu, Allan dos Santos Costa, Maryam Fazel-Zarandi, Tom Sercu, Sal Candido, et al. Language models of protein sequences at the scale of evolution enable accurate structure prediction.BioRxiv, 2022: 500902, 2022
2022
-
[24]
Prottrans: toward understanding the language of life through self-supervised learning.IEEE transactions on pattern analysis and machine intelligence, 44(10):7112–7127, 2021
Ahmed Elnaggar, Michael Heinzinger, Christian Dallago, Ghalia Rehawi, Yu Wang, Llion Jones, Tom Gibbs, Tamas Feher, Christoph Angerer, Martin Steinegger, et al. Prottrans: toward understanding the language of life through self-supervised learning.IEEE transactions on pattern analysis and machine intelligence, 44(10):7112–7127, 2021
2021
-
[25]
Protgo: Function-guided protein modeling for unified representation learning
Bozhen Hu, Cheng Tan, Yongjie Xu, Zhangyang Gao, Jun Xia, Lirong Wu, and Stan Z Li. Protgo: Function-guided protein modeling for unified representation learning. InAdvances in Neural Information Processing Systems, volume 37, pages 88581–88604, 2024
2024
-
[26]
arXiv preprint arXiv:2203.06125 , year=
Zuobai Zhang, Minghao Xu, Arian Jamasb, Vijil Chenthamarakshan, Aurelie Lozano, Payel Das, and Jian Tang. Protein representation learning by geometric structure pretraining.arXiv preprint arXiv:2203.06125, 2022
-
[27]
High-resolution de novo structure prediction from primary sequence.BioRxiv, pages 2022–07, 2022
Ruidong Wu, Fan Ding, Rui Wang, Rui Shen, Xiwen Zhang, Shitong Luo, Chenpeng Su, Zuofan Wu, Qi Xie, Bonnie Berger, et al. High-resolution de novo structure prediction from primary sequence.BioRxiv, pages 2022–07, 2022. 11
2022
-
[28]
Ultrafast and accurate sequence alignment and clustering of viral genomes.Nature Methods, 22(6):1191–1194, 2025
Andrzej Zielezinski, Adam Gudy´s, Jakub Barylski, Krzysztof Siminski, Piotr Rozwalak, Bas E Dutilh, and Sebastian Deorowicz. Ultrafast and accurate sequence alignment and clustering of viral genomes.Nature Methods, 22(6):1191–1194, 2025
2025
-
[29]
Resapred: A deep residual network with self-attention to predict protein flexibility.IEEE Transactions on Computational Biology and Bioinformatics, 22(1):216–227, 2025
Wei Wang, Shitong Wan, Hu Jin, Dong Liu, Hongjun Zhang, Yun Zhou, and Xianfang Wang. Resapred: A deep residual network with self-attention to predict protein flexibility.IEEE Transactions on Computational Biology and Bioinformatics, 22(1):216–227, 2025
2025
-
[30]
Learning to engineer protein flexibility
Petr Kouba et al. Learning to engineer protein flexibility. InInternational Conference on Learning Representations, 2025
2025
-
[31]
Deep-probind: binding protein prediction with transformer- based deep learning model.BMC bioinformatics, 26(1):88, 2025
Salman Khan, Sumaiya Noor, Hamid Hussain Awan, Shehryar Iqbal, Salman A AlQahtani, Naqqash Dilshad, and Nijad Ahmad. Deep-probind: binding protein prediction with transformer- based deep learning model.BMC bioinformatics, 26(1):88, 2025
2025
-
[32]
Mmsite: a multi- modal framework for the identification of active sites in proteins
Song Ouyang, Huiyu Cai, Yong Luo, Kehua Su, Lefei Zhang, and Bo Du. Mmsite: a multi- modal framework for the identification of active sites in proteins. InAdvances in Neural Information Processing Systems, volume 37, pages 45819–45849, 2024
2024
-
[33]
M3site: multiclass multimodal learning for protein active site identification and classification.Briefings in Bioinformatics, 26(6):bbaf590, 2025
Song Ouyang, Yong Luo, Huiyu Cai, Kehua Su, Fei Liao, Na Zhan, Huangxuan Zhao, Tailang Yin, Lin Zhao, and Dongjing Shan. M3site: multiclass multimodal learning for protein active site identification and classification.Briefings in Bioinformatics, 26(6):bbaf590, 2025
2025
-
[34]
Deep learning guided optimization of human antibody against sars-cov-2 variants with broad neutralization.Proceedings of the National Academy of Sciences, 119(11):e2122954119, 2022
Sisi Shan, Shitong Luo, Ziqing Yang, Junxian Hong, Yufeng Su, Fan Ding, Lili Fu, Chenyu Li, Peng Chen, Jianzhu Ma, et al. Deep learning guided optimization of human antibody against sars-cov-2 variants with broad neutralization.Proceedings of the National Academy of Sciences, 119(11):e2122954119, 2022
2022
-
[35]
Pretrainable geometric graph neural network for antibody affinity maturation.Nature Communications, 15(1):7785, 2024
Huiyu Cai, Zuobai Zhang, Mingkai Wang, Bozitao Zhong, Quanxiao Li, Yuxuan Zhong, Yanling Wu, Tianlei Ying, and Jian Tang. Pretrainable geometric graph neural network for antibody affinity maturation.Nature Communications, 15(1):7785, 2024
2024
-
[36]
Ppi-graphomer: enhanced protein-protein affinity prediction using pretrained and graph transformer models.BMC bioinformatics, 26(1):116, 2025
Jun Xie, Youli Zhang, Ziyang Wang, Xiaocheng Jin, Xiaoli Lu, Shengxiang Ge, and Xiaoping Min. Ppi-graphomer: enhanced protein-protein affinity prediction using pretrained and graph transformer models.BMC bioinformatics, 26(1):116, 2025
2025
-
[37]
Island: in-silico proteins binding affinity prediction using sequence information
Wajid Arshad Abbasi, Adiba Yaseen, Fahad Ul Hassan, Saiqa Andleeb, and Fayyaz Ul Amir Af- sar Minhas. Island: in-silico proteins binding affinity prediction using sequence information. BioData Mining, 13(1):20, 2020
2020
-
[38]
Anton Bushuiev, Roman Bushuiev, Petr Kouba, Anatolii Filkin, Marketa Gabrielova, Michal Gabriel, Jiri Sedlar, Tomas Pluskal, Jiri Damborsky, Stanislav Mazurenko, et al. Learning to de- sign protein-protein interactions with enhanced generalization.arXiv preprint arXiv:2310.18515, 2023
-
[39]
Probass—a language model with sequence and structural features for predicting the effect of mutations on binding affinity.Bioinformatics, 41(5):btaf270, 2025
Sagara NS Gurusinghe, Yibing Wu, William DeGrado, and Julia M Shifman. Probass—a language model with sequence and structural features for predicting the effect of mutations on binding affinity.Bioinformatics, 41(5):btaf270, 2025
2025
-
[40]
Dgcddg: deep graph convolution for predicting protein-protein binding affinity changes upon mutations
Yelu Jiang, Lijun Quan, Kailong Li, Yan Li, Yiting Zhou, Tingfang Wu, and Qiang Lyu. Dgcddg: deep graph convolution for predicting protein-protein binding affinity changes upon mutations. IEEE/ACM Transactions on Computational Biology and Bioinformatics, 20(3):2089–2100, 2023
2089
-
[41]
Multi-scale feature fusion network for the prediction of protein-protein binding affinity changes upon mutations
Hao Zhang, Yang Liu, Limin Yu, Zejie Wang, Yifei Liu, and Maozu Guo. Multi-scale feature fusion network for the prediction of protein-protein binding affinity changes upon mutations. In2025 IEEE International Conference on Bioinformatics and Biomedicine (BIBM), pages 218–223. IEEE, 2025
2025
-
[42]
Cryodrgn: reconstruction of heterogeneous cryo-em structures using neural networks.Nature Methods, 18(2):176–185, 2021
Ellen D Zhong, Tristan Bepler, Bonnie Berger, and Joseph H Davis. Cryodrgn: reconstruction of heterogeneous cryo-em structures using neural networks.Nature Methods, 18(2):176–185, 2021. 12
2021
-
[43]
High-resolution real-space reconstruction of cryo-em structures using a neural field network.Nature Machine Intelligence, 6(8):892–903, 2024
Yue Huang, Chengguang Zhu, Xiaokang Yang, and Manhua Liu. High-resolution real-space reconstruction of cryo-em structures using a neural field network.Nature Machine Intelligence, 6(8):892–903, 2024
2024
-
[44]
Emdatabank unified data resource for 3dem.Nucleic acids research, 44(D1):D396–D403, 2016
Catherine L Lawson, Ardan Patwardhan, Matthew L Baker, Corey Hryc, Eduardo Sanz Gar- cia, Brian P Hudson, Ingvar Lagerstedt, Steven J Ludtke, Grigore Pintilie, Raul Sala, et al. Emdatabank unified data resource for 3dem.Nucleic acids research, 44(D1):D396–D403, 2016
2016
-
[45]
Cryotransformer: a trans- former model for picking protein particles from cryo-em micrographs.Bioinformatics, 40(3): btae109, 2024
Ashwin Dhakal, Rajan Gyawali, Liguo Wang, and Jianlin Cheng. Cryotransformer: a trans- former model for picking protein particles from cryo-em micrographs.Bioinformatics, 40(3): btae109, 2024
2024
-
[46]
Emol: modeling protein-nucleic acid complex structures from cryo-em maps by coupling chain assembly with map segmentation.Nucleic acids research, 53(W1):W228–W237, 2025
Ziying Zhang, Liang Xu, Shuai Zhang, Chunxiang Peng, Guijun Zhang, and Xiaogen Zhou. Emol: modeling protein-nucleic acid complex structures from cryo-em maps by coupling chain assembly with map segmentation.Nucleic acids research, 53(W1):W228–W237, 2025
2025
-
[47]
Unlocking de novo antibody design with generative artificial intelligence.BioRxiv, pages 2023–01, 2023
Amir Shanehsazzadeh, Sharrol Bachas, Matt McPartlon, George Kasun, John M Sutton, An- drea K Steiger, Richard Shuai, Christa Kohnert, Goran Rakocevic, Jahir M Gutierrez, et al. Unlocking de novo antibody design with generative artificial intelligence.BioRxiv, pages 2023–01, 2023
2023
-
[48]
Skempi 2.0: an updated benchmark of changes in protein–protein binding energy, kinetics and thermodynamics upon mutation.Bioinformatics, 35(3):462–469, 2019
Justina Jankauskait˙e, Brian Jiménez-García, Justas Dapk¯unas, Juan Fernández-Recio, and Iain H Moal. Skempi 2.0: an updated benchmark of changes in protein–protein binding energy, kinetics and thermodynamics upon mutation.Bioinformatics, 35(3):462–469, 2019
2019
-
[49]
Proteinnet: a standardized data set for machine learning of protein structure.BMC bioinformatics, 20(1):311, 2019
Mohammed AlQuraishi. Proteinnet: a standardized data set for machine learning of protein structure.BMC bioinformatics, 20(1):311, 2019
2019
-
[50]
Cryp- tobench: cryptic protein–ligand binding sites dataset and benchmark.Bioinformatics, 41(1): btae745, 2025
Vít Škrhák, Marian Novotn `y, Christos P Feidakis, Radoslav Krivák, and David Hoksza. Cryp- tobench: cryptic protein–ligand binding sites dataset and benchmark.Bioinformatics, 41(1): btae745, 2025
2025
-
[51]
Learning inverse folding from millions of predicted structures
Chloe Hsu, Robert Verkuil, Jason Liu, Zeming Lin, Brian Hie, Tom Sercu, Adam Lerer, and Alexander Rives. Learning inverse folding from millions of predicted structures. InInternational Conference on Machine Learning, pages 8946–8970. PMLR, 2022
2022
-
[52]
Controllable protein design with language models.Nature Machine Intelligence, 4(6):521–532, 2022
Noelia Ferruz and Birte Höcker. Controllable protein design with language models.Nature Machine Intelligence, 4(6):521–532, 2022
2022
-
[53]
Gomez, Łukasz Kaiser, and Illia Polosukhin
Ashish Vaswani, Noam Shazeer, Niki Parmar, Jakob Uszkoreit, Llion Jones, Aidan N. Gomez, Łukasz Kaiser, and Illia Polosukhin. Attention is all you need. InAdvances in Neural Informa- tion Processing Systems, volume 30, 2017
2017
-
[54]
Aixin Liu, Bei Feng, Bing Xue, Bingxuan Wang, Bochao Wu, Chengda Lu, Chenggang Zhao, Chengqi Deng, Chenyu Zhang, Chong Ruan, et al. Deepseek-v3 technical report.arXiv preprint arXiv:2412.19437, 2024
work page internal anchor Pith review Pith/arXiv arXiv 2024
-
[55]
Multi-to-uni modal knowledge transfer pre-training for molecular representation learning.Nature Communications, 2026
Zhankun Xiong, Ziyan Wang, Feng Huang, Minyao Qiu, Shuyan Fang, Liuqing Yang, Xionghui Zhou, Shichao Liu, Ping Zhang, and Wen Zhang. Multi-to-uni modal knowledge transfer pre-training for molecular representation learning.Nature Communications, 2026
2026
-
[56]
Unsupervised domain adaptation by backpropagation
Yaroslav Ganin and Victor Lempitsky. Unsupervised domain adaptation by backpropagation. InInternational Conference on Machine Learning, pages 1180–1189. PMLR, 2015
2015
-
[57]
Multi- modal learning with missing modality via shared-specific feature modelling
Hu Wang, Yuanhong Chen, Congbo Ma, Jodie Avery, Louise Hull, and Gustavo Carneiro. Multi- modal learning with missing modality via shared-specific feature modelling. InProceedings of the IEEE/CVF conference on computer vision and pattern recognition, pages 15878–15887, 2023
2023
-
[58]
Multi-task learning using uncertainty to weigh losses for scene geometry and semantics
Alex Kendall, Yarin Gal, and Roberto Cipolla. Multi-task learning using uncertainty to weigh losses for scene geometry and semantics. InProceedings of the IEEE conference on computer vision and pattern recognition, pages 7482–7491, 2018. 13
2018
-
[59]
What uncertainties do we need in bayesian deep learning for computer vision? InAdvances in Neural Information Processing Systems, volume 30, 2017
Alex Kendall and Yarin Gal. What uncertainties do we need in bayesian deep learning for computer vision? InAdvances in Neural Information Processing Systems, volume 30, 2017
2017
-
[60]
High-resolution cryo-em of the human cdk-activating kinase for structure-based drug design
Victoria I Cushing, Adrian F Koh, Junjie Feng, Kaste Jurgaityte, Alexander Bondke, Sebas- tian HB Kroll, Marion Barbazanges, Bodo Scheiper, Ash K Bahl, Anthony GM Barrett, et al. High-resolution cryo-em of the human cdk-activating kinase for structure-based drug design. Nature Communications, 15(1):2265, 2024
2024
-
[61]
Measuring local-directional resolution and local anisotropy in cryo-em maps.Nature Communications, 11(1):55, 2020
Jose Luis Vilas, Hemant D Tagare, Javier Vargas, Jose Maria Carazo, and Carlos Oscar S Sorzano. Measuring local-directional resolution and local anisotropy in cryo-em maps.Nature Communications, 11(1):55, 2020
2020
-
[62]
Automatic local resolution-based sharpening of cryo-em maps.Bioinformatics, 36(3):765–772, 2020
Erney Ramírez-Aportela, Jose Luis Vilas, Alisa Glukhova, Roberto Melero, Pablo Conesa, Marta Martínez, David Maluenda, Javier Mota, Amaya Jiménez, Javier Vargas, et al. Automatic local resolution-based sharpening of cryo-em maps.Bioinformatics, 36(3):765–772, 2020
2020
-
[63]
Respre: high-accuracy protein contact prediction by coupling precision matrix with deep residual neural networks
Yang Li, Jun Hu, Chengxin Zhang, Dong-Jun Yu, and Yang Zhang. Respre: high-accuracy protein contact prediction by coupling precision matrix with deep residual neural networks. Bioinformatics, 35(22):4647–4655, 2019
2019
-
[64]
A deep learning framework for improving long-range residue–residue contact prediction using a hierarchical strategy.Bioinformatics, 33 (17):2675–2683, 2017
Dapeng Xiong, Jianyang Zeng, and Haipeng Gong. A deep learning framework for improving long-range residue–residue contact prediction using a hierarchical strategy.Bioinformatics, 33 (17):2675–2683, 2017. 14 Appendix A Dataset Details Pretraining dataset.The pretraining data are collected from EMDB [ 44], which provides a large number of experimentally det...
2017
-
[65]
represents protein–protein interfaces as all-atom graphs and models interactions through multi- level geometric message passing, enabling effective characterization of complex intermolecular interactions.Island[ 37] is a sequence-driven approach for binding affinity prediction. It utilizes a variety of features derived from protein sequences and applies r...
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.