scTranslation: A Comprehensive Benchmark for Single-Cell Multi-Omics Modality Translation
Pith reviewed 2026-06-28 10:01 UTC · model grok-4.3
The pith
scTranslation benchmark enables systematic study of single-cell multi-omics modality translation methods and their performance factors.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
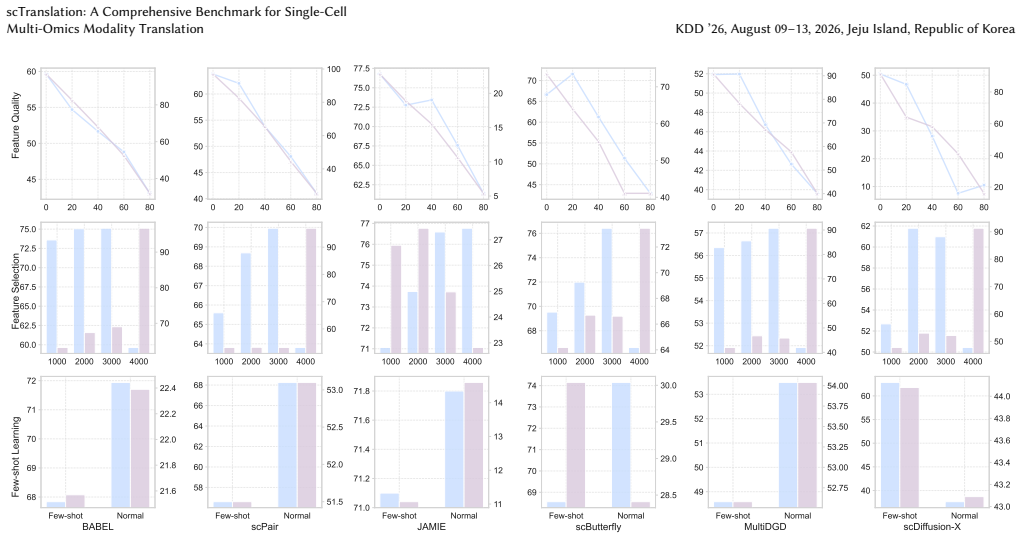
We present scTranslation, a comprehensive benchmark for single-cell multi-omics modality translation tasks. It includes diverse translation datasets, integrates state-of-the-art models, and provides a comprehensive evaluation metrics. In addition, we assess model performance under different scenarios, such as feature selection, feature quality, and few-shot settings. Leveraging this benchmark, we conduct a large-scale study of current methods, report many insightful findings that open up new possibilities for future development.
What carries the argument
scTranslation benchmark, which organizes datasets, models, and metrics to enable controlled comparisons of modality translation performance.
If this is right
- Model performance changes substantially with different feature selection strategies.
- Few-shot settings expose weaknesses in existing translation approaches.
- Feature quality directly influences translation accuracy.
- The benchmark supports reproducible large-scale comparisons.
- Open-sourcing the code allows others to build on the findings.
Where Pith is reading between the lines
- Future work could expand the benchmark with additional real-world datasets to test generalizability.
- Developers might prioritize methods that handle low-quality features well based on these results.
- The findings could help prioritize which modalities to measure experimentally when resources are limited.
Load-bearing premise
The datasets, models, and metrics chosen for the benchmark are representative and unbiased for drawing general conclusions about modality translation.
What would settle it
If re-running the study with a substantially different set of datasets or metrics leads to contradictory rankings of the methods, that would indicate the benchmark does not support reliable general conclusions.
Figures


read the original abstract
Simultaneous measurement of multiple omics modalities in single cells enables researchers to gain a more comprehensive understanding of cellular states and regulatory mechanisms. However, due to high experimental costs, significant noise, and incomplete modality coverage, a variety of computational methods for modality translation have emerged in recent years. Despite the development of translation models, there is still a lack of systematic benchmark evaluation in terms of datasets, evaluation metrics, and influencing factors. To address this, we present scTranslation, a comprehensive benchmark for single-cell multi-omics modality translation tasks. It includes diverse translation datasets, integrates state-of-the-art models, and provides a comprehensive evaluation metrics. In addition, we assess model performance under different scenarios, such as feature selection, feature quality, and few-shot settings. These factors significantly affect model performance but have rarely been systematically studied before. Leveraging this benchmark, we conduct a large-scale study of current methods, report many insightful findings that open up new possibilities for future development. The benchmark is open-sourced to facilitate future research. The code is anonymously released at https://github.com/Bunnybeibei/scTranslation.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript introduces scTranslation, a benchmark for single-cell multi-omics modality translation. It includes diverse translation datasets, integrates state-of-the-art models, provides comprehensive evaluation metrics, and assesses performance under scenarios such as feature selection, feature quality, and few-shot settings. The authors conduct a large-scale study of current methods and report insightful findings to guide future development; the benchmark and code are released openly.
Significance. If the datasets, models, and metrics are representative and the evaluation protocols are sound, the benchmark could standardize comparisons in single-cell modality translation and highlight under-studied factors, thereby supporting more reliable method development in the field.
minor comments (3)
- The abstract states that the benchmark includes 'diverse translation datasets' and 'comprehensive evaluation metrics' but does not specify selection criteria or exact metric definitions; these details should be expanded in §2 or §3 with explicit lists and justifications.
- The claim of 'many insightful findings' is asserted without previewing any concrete examples (e.g., performance rankings or scenario-specific trends); adding a brief summary of key results in the abstract or introduction would improve clarity.
- The GitHub link is provided, but the manuscript does not indicate whether the released code includes the exact dataset splits, preprocessing scripts, and statistical test implementations used in the reported experiments.
Simulated Author's Rebuttal
We thank the referee for their positive summary of the scTranslation benchmark, including its datasets, integrated models, evaluation metrics, and analysis under feature selection, data quality, and few-shot scenarios. We note the recommendation for major revision but observe that the report contains no specific major comments to address point by point. We remain available to incorporate any additional feedback from the editor or referee.
Circularity Check
No significant circularity; empirical benchmark with no derivation chain
full rationale
The paper is a benchmark study introducing datasets, integrating SOTA models, and evaluating under various scenarios (feature selection, few-shot, etc.). No equations, fitted parameters, predictions, or uniqueness theorems are present. The central claim—that the benchmark enables insightful findings—rests on standard empirical construction rather than any self-referential reduction. No load-bearing self-citations or ansatzes are invoked. This is a self-contained empirical contribution with no circular steps.
Axiom & Free-Parameter Ledger
axioms (2)
- domain assumption The collected datasets are diverse and representative of single-cell multi-omics modality translation tasks.
- domain assumption The integrated state-of-the-art models and chosen evaluation metrics are appropriate for assessing translation performance.
Reference graph
Works this paper leans on
-
[1]
Ricard Argelaguet, Damien Arnol, Danila Bredikhin, Yonatan Deloro, Britta Velten, John C Marioni, and Oliver Stegle. 2020. MOFA+: a statistical framework for comprehensive integration of multi-modal single-cell data.Genome biology 21, 1 (2020), 111
2020
-
[2]
Ricard Argelaguet, Anna SE Cuomo, Oliver Stegle, and John C Marioni. 2021. Computational principles and challenges in single-cell data integration.Nature biotechnology39, 10 (2021), 1202–1215
2021
-
[3]
Ricard Argelaguet, Britta Velten, Damien Arnol, Sascha Dietrich, Thorsten Zenz, John C Marioni, Florian Buettner, Wolfgang Huber, and Oliver Stegle. 2018. Multi- Omics Factor Analysis—a framework for unsupervised integration of multi-omics data sets.Molecular systems biology14, 6 (2018), MSB178124
2018
-
[4]
Tal Ashuach, Mariano I Gabitto, Rohan V Koodli, Giuseppe-Antonio Saldi, Michael I Jordan, and Nir Yosef. 2023. MultiVI: deep generative model for the integration of multimodal data.Nature methods20, 8 (2023), 1222–1231
2023
-
[5]
Tasbiraha Athaya, Rony Chowdhury Ripan, Xiaoman Li, and Haiyan Hu. 2023. Multimodal deep learning approaches for single-cell multi-omics data integration. Briefings in bioinformatics24, 5 (2023), bbad313
2023
-
[6]
Alev Baysoy, Zhiliang Bai, Rahul Satija, and Rong Fan. 2023. The technological landscape and applications of single-cell multi-omics.Nature Reviews Molecular Cell Biology24, 10 (2023), 695–713
2023
-
[7]
Christoph Bock, Matthias Farlik, and Nathan C Sheffield. 2016. Multi-omics of single cells: strategies and applications.Trends in biotechnology34, 8 (2016), 605–608
2016
-
[8]
Jason D Buenrostro, Alexander Corces, Cara A Lareau, Brian Wu, Allison N Schep, Martin J Aryee, Ravindra Majeti, Howard Y Chang, and William J Greenleaf
-
[9]
Nature523, 7561 (2015), 486–490
Single-cell chromatin accessibility reveals principles of regulatory variation. Nature523, 7561 (2015), 486–490
2015
-
[10]
Jason D Buenrostro, Paul G Giresi, Lisa C Zaba, Howard Y Chang, and William J Greenleaf. 2013. Transposition of native chromatin for fast and sensitive epige- nomic profiling of open chromatin, DNA-binding proteins and nucleosome posi- tion.Nature methods10, 12 (2013), 1213–1218
2013
-
[11]
Junyue Cao, Darren A Cusanovich, Vijay Ramani, Delasa Aghamirzaie, Hannah A Pliner, Andrew J Hill, Riza M Daza, Jose L McFaline-Figueroa, Jonathan S Packer, Lena Christiansen, et al. 2018. Joint profiling of chromatin accessibility and gene expression in thousands of single cells.Science361, 6409 (2018), 1380–1385
2018
-
[12]
Yichuan Cao, Xiamiao Zhao, Songming Tang, Qun Jiang, Sijie Li, Siyu Li, and Shengquan Chen. 2024. scButterfly: a versatile single-cell cross-modality transla- tion method via dual-aligned variational autoencoders.Nature Communications 15, 1 (2024), 2973
2024
-
[13]
Zhi-Jie Cao and Ge Gao. 2022. Multi-omics single-cell data integration and regulatory inference with graph-linked embedding.Nature biotechnology40, 10 (2022), 1458–1466
2022
-
[14]
Song Chen, Blue B Lake, and Kun Zhang. 2019. High-throughput sequencing of the transcriptome and chromatin accessibility in the same cell.Nature biotech- nology37, 12 (2019), 1452–1457
2019
-
[15]
Stephen J Clark, Ricard Argelaguet, Chantriolnt-Andreas Kapourani, Thomas M Stubbs, Heather J Lee, Celia Alda-Catalinas, Felix Krueger, Guido Sanguinetti, Gavin Kelsey, John C Marioni, et al . 2018. scNMT-seq enables joint profiling of chromatin accessibility DNA methylation and transcription in single cells. Nature communications9, 1 (2018), 781
2018
-
[16]
Noah Cohen Kalafut, Xiang Huang, and Daifeng Wang. 2023. Joint variational autoencoders for multimodal imputation and embedding.Nature machine intelli- gence5, 6 (2023), 631–642
2023
-
[17]
Damian A Cusanovich, Amanda J Hill, Daria Aghamirzaie, Ruben M Daza, Han- nah A Pliner, Justin B Berletch, Galina N Filippova, Xinxin Huang, Lasse Chris- tiansen, William S DeWitt, et al. 2018. A single-cell atlas of in vivo mammalian chromatin accessibility.Cell174, 5 (2018), 1309–1324
2018
-
[18]
Atray Dixit, Oren Parnas, Biyu Li, Jenny Chen, Charles P Fulco, Livnat Jerby- Arnon, Nemanja D Marjanovic, Danielle Dionne, Tyler Burks, Raktima Ray- chowdhury, et al. 2016. Perturb-Seq: dissecting molecular circuits with scalable single-cell RNA profiling of pooled genetic screens.cell167, 7 (2016), 1853–1866
2016
-
[19]
Jin-Hong Du, Zhanrui Cai, and Kathryn Roeder. 2022. Robust probabilistic mod- eling for single-cell multimodal mosaic integration and imputation via scVAEIT. Proceedings of the National Academy of Sciences119, 49 (2022), e2214414119
2022
-
[20]
Adam Gayoso, Zoë Steier, Romain Lopez, Jeffrey Regier, Kristopher L Nazor, Aaron Streets, and Nir Yosef. 2021. Joint probabilistic modeling of single-cell multi-omic data with totalVI.Nature methods18, 3 (2021), 272–282
2021
-
[21]
Boying Gong, Yun Zhou, and Elizabeth Purdom. 2021. Cobolt: integrative analysis of multimodal single-cell sequencing data.Genome biology22, 1 (2021), 351
2021
-
[22]
Jonathan Ho, Ajay Jain, and Pieter Abbeel. 2020. Denoising diffusion probabilistic models.Advances in neural information processing systems33 (2020), 6840–6851
2020
-
[23]
Hongru Hu and Gerald Quon. 2024. scPair: Boosting single cell multimodal analysis by leveraging implicit feature selection and single cell atlases.Nature Communications15, 1 (2024), 9932
2024
-
[24]
Mohammed Inayatullah, Anuj Kumar Dwivedi, and Vijay K Tiwari. 2025. Ad- vances in single-cell omics: Transformative applications in basic and clinical research.Current Opinion in Cell Biology95 (2025), 102548
2025
-
[25]
Julia Källberg, Wenjin Xiao, David Van Assche, Jean-Christophe Baret, and Valerie Taly. 2022. Frontiers in single cell analysis: multimodal technologies and their clinical perspectives.Lab on a Chip22, 13 (2022), 2403–2422
2022
-
[26]
Diederik P Kingma and Max Welling. 2013. Auto-encoding variational bayes. arXiv preprint arXiv:1312.6114(2013)
work page internal anchor Pith review Pith/arXiv arXiv 2013
-
[27]
Pengzhi Li, Yan Pei, and Jianqiang Li. 2023. A comprehensive survey on design and application of autoencoder in deep learning.Applied Soft Computing138 (2023), 110176
2023
-
[28]
Jongsu Lim, Chanho Park, Minjae Kim, Hyukhee Kim, Junil Kim, and Dong-Sung Lee. 2024. Advances in single-cell omics and multiomics for high-resolution molecular profiling.Experimental & molecular medicine56, 3 (2024), 515–526
2024
-
[29]
Longqi Liu, Chuanyu Liu, Andrés Quintero, Liang Wu, Yue Yuan, Mingyue Wang, Mengnan Cheng, Lizhi Leng, Liqin Xu, Guoyi Dong, et al . 2019. Deconvolu- tion of single-cell multi-omics layers reveals regulatory heterogeneity.Nature communications10, 1 (2019), 470
2019
-
[30]
Romain Lopez, Jeffrey Regier, Michael B Cole, Michael I Jordan, and Nir Yosef
-
[31]
Deep generative modeling for single-cell transcriptomics.Nature methods 15, 12 (2018), 1053–1058
2018
-
[32]
Mohammad Lotfollahi, F Alexander Wolf, and Fabian J Theis. 2019. scGen predicts single-cell perturbation responses.Nature methods16, 8 (2019), 715–721
2019
-
[33]
Malte D Luecken, Daniel Bernard Burkhardt, Robrecht Cannoodt, Christopher Lance, Aditi Agrawal, Hananeh Aliee, Ann T Chen, Louise Deconinck, Angela M Detweiler, Alejandro A Granados, et al . 2021. A sandbox for prediction and integration of DNA, RNA, and proteins in single cells. In35th conference on neural information processing systems (NeurIPS 2021) tr...
2021
-
[34]
Erpai Luo, Minsheng Hao, Lei Wei, and Xuegong Zhang. 2024. scDiffusion: conditional generation of high-quality single-cell data using diffusion model. Bioinformatics40, 9 (2024), btae518
2024
-
[35]
Zhen Lyu, Sabin Dahal, Shuai Zeng, Juexin Wang, Dong Xu, and Trupti Joshi
-
[36]
CrossMP: Enabling Cross-Modality Translation between Single-Cell RNA- Seq and Single-Cell ATAC-Seq through Web-Based Portal.Genes15, 7 (2024), 882
2024
-
[37]
Aviv Regev, Sarah A Teichmann, Eric S Lander, Ido Amit, Christophe Benoist, Ewan Birney, Bernd Bodenmiller, Peter Campbell, Piero Carninci, Menna Clat- worthy, et al. 2017. The human cell atlas.elife6 (2017), e27041
2017
-
[38]
Douglas Reynolds. 2015. Gaussian mixture models. InEncyclopedia of biometrics. Springer, 827–832
2015
-
[39]
Viktoria Schuster, Emma Dann, Anders Krogh, and Sarah A Teichmann. 2024. multiDGD: A versatile deep generative model for multi-omics data.Nature Communications15, 1 (2024), 10031
2024
-
[40]
Harrison Specht, Edward Emmott, Aleksandra A Petelski, R Gray Huffman, David H Perlman, Marco Serra, Peter Kharchenko, Antonius Koller, and Nikolai Slavov. 2021. Single-cell proteomic and transcriptomic analysis of macrophage heterogeneity using SCoPE2.Genome biology22, 1 (2021), 50
2021
-
[41]
Marlon Stoeckius, Christoph Hafemeister, William Stephenson, Brian Houck- Loomis, Pratip K Chattopadhyay, Harold Swerdlow, Rahul Satija, and Peter Smib- ert. 2017. Simultaneous epitope and transcriptome measurement in single cells. Nature methods14, 9 (2017), 865–868
2017
-
[42]
Tim Stuart, Andrew Butler, Paul Hoffman, Christoph Hafemeister, Efthymia Papalexi, William M Mauck, Yuhan Hao, Marlon Stoeckius, Peter Smibert, and Rahul Satija. 2019. Comprehensive integration of single-cell data.cell177, 7 (2019), 1888–1902
2019
-
[43]
Indhupriya Subramanian, Srikant Verma, Shiva Kumar, Abhay Jere, and Kris- hanpal Anamika. 2020. Multi-omics data integration, interpretation, and its application.Bioinformatics and biology insights14 (2020), 1177932219899051
2020
-
[44]
Fuchou Tang, Catalin Barbacioru, Yangzhou Wang, Ellen Nordman, Clarence Lee, Nanlan Xu, Xiaohui Wang, John Bodeau, Brian B Tuch, Asim Siddiqui, et al. 2009. mRNA-Seq whole-transcriptome analysis of a single cell.Nature methods6, 5 (2009), 377–382
2009
-
[45]
Alexandro E Trevino, Fabian Müller, Jimena Andersen, Laksshman Sundaram, Arwa Kathiria, Anna Shcherbina, Kyle Farh, Howard Y Chang, Anca M Pas,ca, Anshul Kundaje, et al. 2021. Chromatin and gene-regulatory dynamics of the developing human cerebral cortex at single-cell resolution.Cell184, 19 (2021), 5053–5069
2021
-
[46]
Kevin E Wu, Kathryn E Yost, Howard Y Chang, and James Zou. 2021. BABEL en- ables cross-modality translation between multiomic profiles at single-cell resolu- tion.Proceedings of the National Academy of Sciences118, 15 (2021), e2023070118
2021
-
[47]
Chuxi Xiao, Yixin Chen, Qiuchen Meng, Lei Wei, and Xuegong Zhang. 2024. Benchmarking multi-omics integration algorithms across single-cell RNA and ATAC data.Briefings in Bioinformatics25, 2 (2024), bbae095
2024
-
[48]
Ran Zhang, Laetitia Meng-Papaxanthos, Jean-Philippe Vert, and William Stafford Noble. 2022. Semi-supervised single-cell cross-modality translation using polar- bear. InInternational Conference on Research in Computational Molecular Biology. Springer, 20–35. scTranslation: A Comprehensive Benchmark for Single-Cell Multi-Omics Modality Translation KDD ’26, ...
2022
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.