Beyond Prompt-Based Planning: MCP-Native Graph Planning-based Biomedical Agent System
Pith reviewed 2026-06-28 06:20 UTC · model grok-4.3
The pith
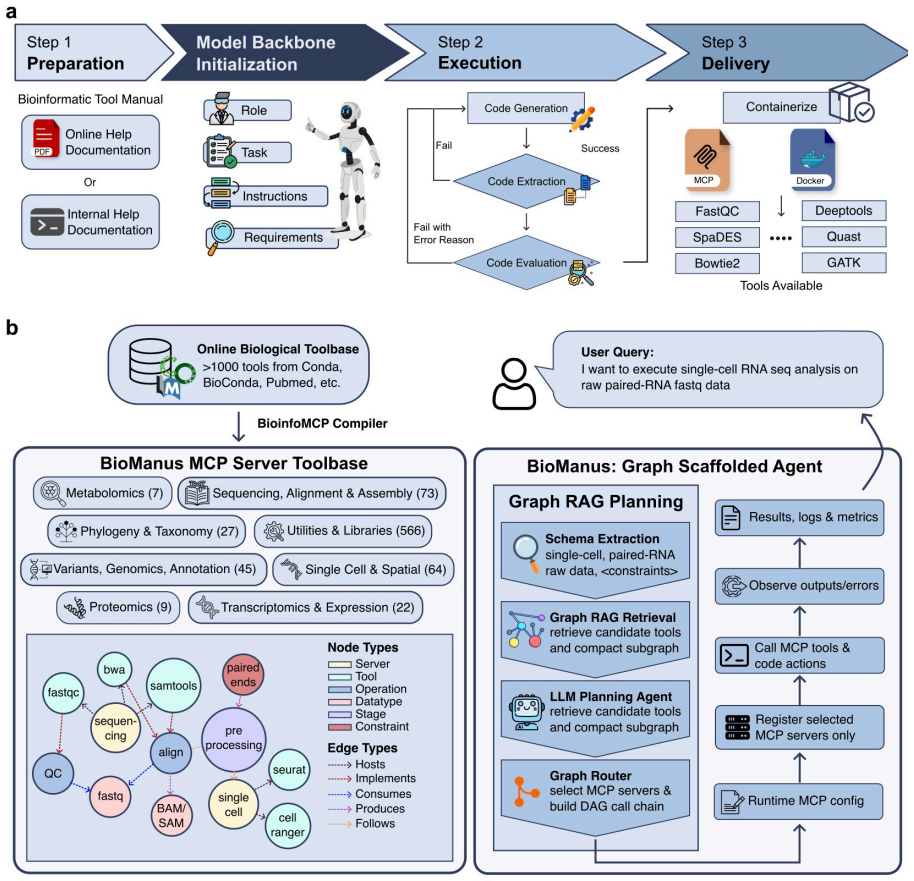
BioManus converts heterogeneous tools into MCP servers, builds a typed graph over them, and retrieves task-specific subgraphs for planning instead of loading every tool description into the prompt.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
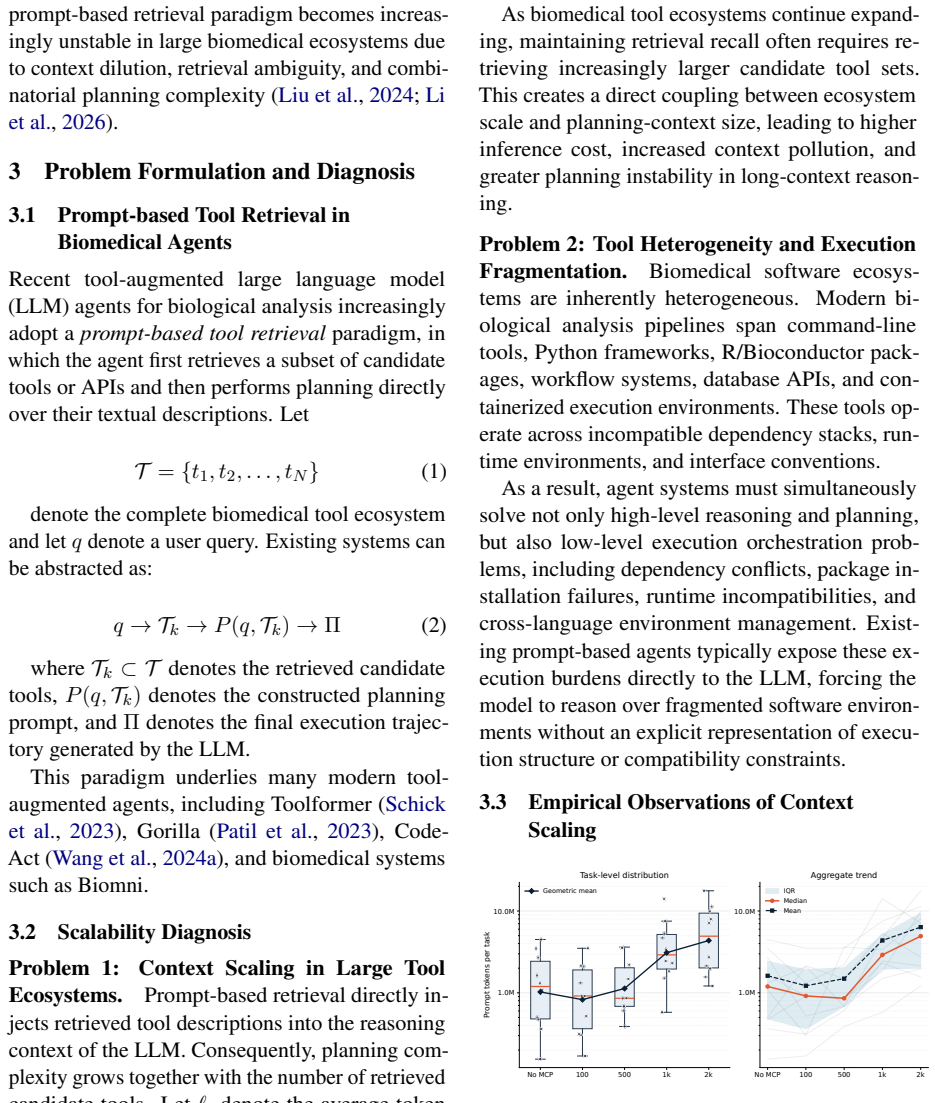
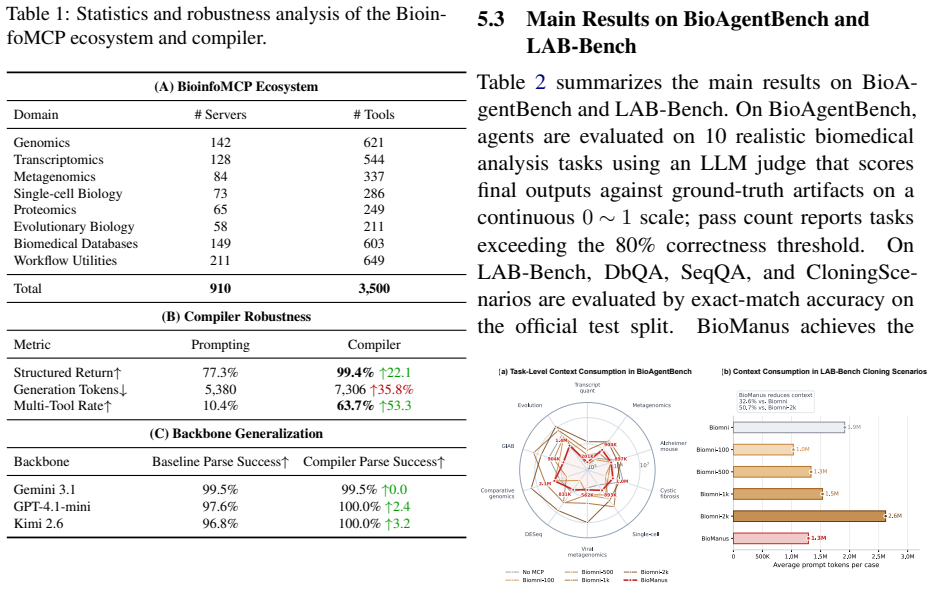
BioManus first introduces the BioinfoMCP Compiler, which converts heterogeneous bioinformatics software into standardized MCP servers, yielding a large executable MCP ecosystem. It then organizes this ecosystem as a typed heterogeneous MCP graph over tools, operations, datatypes, and workflow stages. At inference time, BioManus retrieves compact task-specific subgraphs and synthesizes operation-level workflow scaffolds. This design decouples planning complexity from raw tool inventory size, achieving a context compression ratio of Theta(N / (h * m_bar)) under high-recall retrieval, where N is the total tool count, h is the workflow horizon, and m_bar (much smaller than N) is the average numb
What carries the argument
Typed heterogeneous MCP graph over tools, operations, datatypes, and workflow stages that supports high-recall subgraph retrieval for workflow scaffold synthesis.
Load-bearing premise
Heterogeneous bioinformatics tools can be converted into standardized MCP servers while preserving full functionality and enabling a typed graph that supports high-recall subgraph retrieval without loss of planning quality.
What would settle it
A head-to-head test on the same tool inventory where full prompt-based retrieval produces higher execution accuracy or workflow validity than subgraph retrieval from the MCP graph.
Figures

read the original abstract
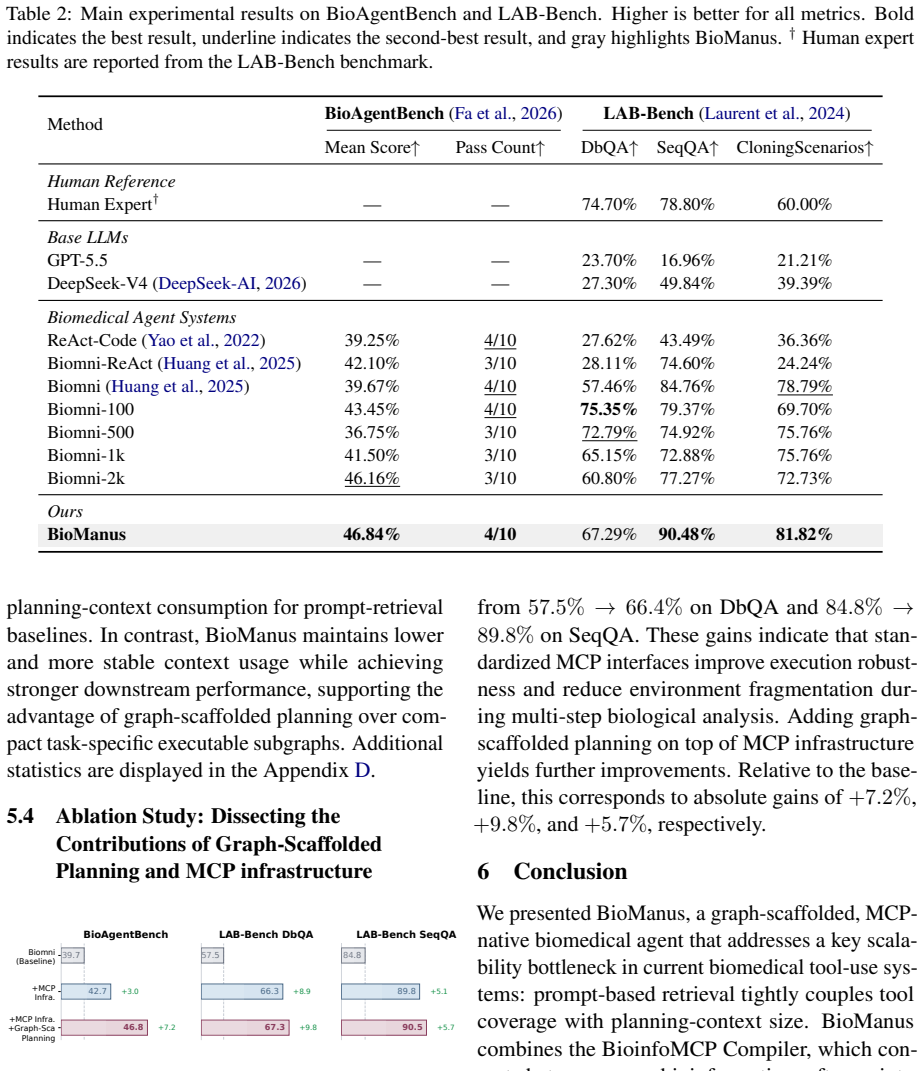
Biomedical agents promise to automate complex biological workflows, yet current systems face two fundamental bottlenecks: bioinformatics tools are highly heterogeneous in interfaces and execution environments, while agent planning still relies on flat prompt-retrieved tool descriptions. As biomedical software ecosystems grow, this coupling between tool coverage and context size leads to tool confusion, unstable planning, and inefficient execution. We introduce BioManus, an MCP-native biomedical agent built on graph-scaffolded planning over structured biological capabilities. BioManus first introduces the BioinfoMCP Compiler, which converts heterogeneous bioinformatics software into standardized MCP servers, yielding a large executable MCP ecosystem. It then organizes this ecosystem as a typed heterogeneous MCP graph over tools, operations, datatypes, and workflow stages. At inference time, BioManus retrieves compact task-specific subgraphs, synthesizes operation-level workflow scaffolds. This design decouples planning complexity from raw tool inventory size, achieving a context compression ratio of Theta(N / (h * m_bar)) under high-recall retrieval, where N is the total tool count, h is the workflow horizon, and m_bar (much smaller than N) is the average number of candidate tools per operation. Experiments on BioAgentBench and LAB-Bench show that BioManus improves execution accuracy, workflow validity, and context efficiency over advanced biomedical agent baselines. This work suggests a paradigm shift: scalable biomedical reasoning requires structured executable capability graphs rather than increasingly larger prompt-level tool retrieval.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The paper introduces BioManus, an MCP-native biomedical agent system. It proposes the BioinfoMCP Compiler to convert heterogeneous bioinformatics tools into standardized MCP servers, organizes the resulting ecosystem as a typed heterogeneous MCP graph over tools/operations/datatypes/workflow stages, performs high-recall subgraph retrieval at inference time to synthesize operation-level workflow scaffolds, and claims a context compression ratio of Theta(N / (h * m_bar)) together with gains in execution accuracy, workflow validity, and context efficiency over baselines on BioAgentBench and LAB-Bench.
Significance. If the compiler successfully standardizes tools while preserving functionality and the typed graph enables high-recall retrieval without degrading planning quality, the work would offer a concrete mechanism for decoupling agent planning complexity from raw tool inventory size. This addresses a recognized scalability bottleneck in biomedical agents and could influence future designs that favor structured executable capability graphs over flat prompt retrieval.
major comments (3)
- [Abstract] Abstract: the central empirical claims (improved execution accuracy, workflow validity, and context efficiency on BioAgentBench and LAB-Bench) are asserted without any description of the experimental protocol, baseline systems, number of runs, error bars, or statistical tests, rendering the performance improvements impossible to evaluate.
- [Abstract] Abstract: the BioinfoMCP Compiler is presented as the load-bearing component that converts heterogeneous tools into standardized MCP servers while preserving full functionality, yet no conversion examples, preservation metrics, or failure modes are supplied; this directly underpins the weakest assumption and the claimed compression ratio.
- [Abstract] Abstract: the compression ratio Theta(N / (h * m_bar)) is stated as a theoretical consequence of the graph design under high-recall retrieval, but no derivation, proof sketch, or explicit relation to the typed-graph construction is provided, leaving open whether the bound holds once retrieval recall and subgraph synthesis costs are accounted for.
minor comments (1)
- [Abstract] The symbol m_bar is introduced without an explicit definition in the abstract sentence that presents the compression formula.
Simulated Author's Rebuttal
We thank the referee for the constructive comments on the abstract. We address each point below and note that the full manuscript contains the supporting details referenced in the responses.
read point-by-point responses
-
Referee: [Abstract] Abstract: the central empirical claims (improved execution accuracy, workflow validity, and context efficiency on BioAgentBench and LAB-Bench) are asserted without any description of the experimental protocol, baseline systems, number of runs, error bars, or statistical tests, rendering the performance improvements impossible to evaluate.
Authors: The abstract is space-constrained and summarizes results whose full protocol, baselines (including advanced biomedical agents), run counts, error bars, and statistical tests appear in the Experiments section of the manuscript. We will revise the abstract to include a concise reference to the evaluation methodology and benchmarks. revision: partial
-
Referee: [Abstract] Abstract: the BioinfoMCP Compiler is presented as the load-bearing component that converts heterogeneous tools into standardized MCP servers while preserving full functionality, yet no conversion examples, preservation metrics, or failure modes are supplied; this directly underpins the weakest assumption and the claimed compression ratio.
Authors: Conversion examples, preservation metrics, and failure-mode analysis for the BioinfoMCP Compiler are supplied in Section 3 of the full manuscript. The abstract introduces the component at a high level; we will add a brief clause noting that functionality preservation was validated. revision: partial
-
Referee: [Abstract] Abstract: the compression ratio Theta(N / (h * m_bar)) is stated as a theoretical consequence of the graph design under high-recall retrieval, but no derivation, proof sketch, or explicit relation to the typed-graph construction is provided, leaving open whether the bound holds once retrieval recall and subgraph synthesis costs are accounted for.
Authors: The derivation of the context compression ratio, its explicit dependence on the typed heterogeneous MCP graph, high-recall subgraph retrieval, and a proof sketch that incorporates retrieval and synthesis costs are given in Section 4 of the manuscript. We will revise the abstract to indicate that the bound is formally derived in the main text. revision: partial
Circularity Check
No significant circularity; derivation self-contained
full rationale
The claimed context compression ratio Theta(N / (h * m_bar)) is presented as a direct mathematical consequence of the graph design's high-recall subgraph retrieval, where context size is bounded by h operations each with m_bar candidates instead of the full N tools. This follows from the explicit definitions of the retrieval mechanism and variables without reducing to a fitted input, self-definition, or self-citation chain. No load-bearing uniqueness theorems, ansatzes via citation, or renamings of known results appear in the abstract or derivation steps. The result is independent of its inputs and does not require external verification to stand as a structural property of the proposed architecture.
Axiom & Free-Parameter Ledger
axioms (1)
- domain assumption Heterogeneous bioinformatics software can be wrapped into standardized MCP servers without loss of capability
invented entities (1)
-
typed heterogeneous MCP graph
no independent evidence
Reference graph
Works this paper leans on
-
[1]
Ruofan Jin, Zaixi Zhang, Mengdi Wang, and Le Cong
Genegpt: augmenting large language models with domain tools for improved access to biomedical information.Bioinformatics, 40(2). Ruofan Jin, Zaixi Zhang, Mengdi Wang, and Le Cong
-
[2]
Stella: Self-evolving llm agent for biomedical research.arXiv preprint arXiv:2507.02004. Johannes Köster and Sven Rahmann. 2012. Snake- make—a scalable bioinformatics workflow engine. Bioinformatics, 28(19):2520–2522. Esther Landhuis. 2016. Scientific literature: Informa- tion overload.Nature, 535(7612):457–458. Jon M. Laurent, Joseph D. Janizek, Michael ...
-
[3]
Gorilla: Large Language Model Connected with Massive APIs
Bioagents: Bridging the gap in bioinformatics analysis with multi-agent systems.Scientific Reports, 15(1):39036. Yi Nian Niu, Eric G Roberts, Danielle Denisko, and Michael M Hoffman. 2022. Assessing and assuring interoperability of a genomics file format.Bioinfor- matics, 38(13):3327–3336. Shishir G. Patil, Tianjun Zhang, Xin Wang, and Joseph E. Gonzalez....
work page internal anchor Pith review Pith/arXiv arXiv 2022
-
[4]
Reproducible, scalable, and shareable analy- sis pipelines with bioinformatics workflow managers. Nature methods, 18(10):1161–1168. Yihang Xiao, Jinyi Liu, Yan Zheng, Xiaohan Xie, Jianye Hao, Mingzhi Li, Ruitao Wang, Fei Ni, Yuxiao Li, Jintian Luo, Shaoqing Jiao, and Jiajie Peng. 2024. Cellagent: An llm-driven multi-agent framework for automated single-ce...
-
[5]
Plan at the operation level before selecting concrete tools.,→
-
[6]
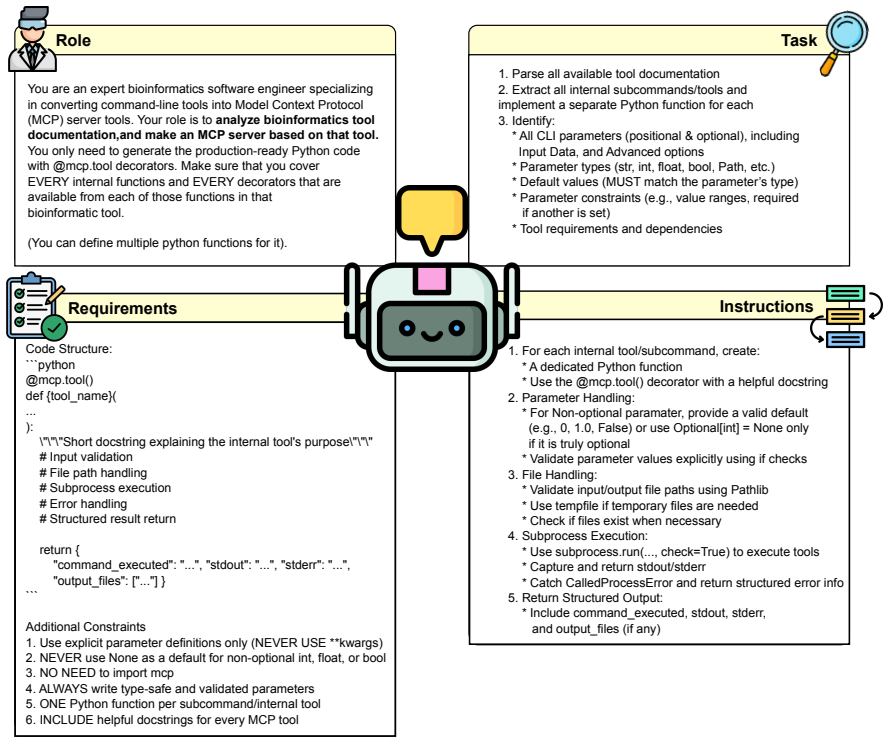
Use only tools appearing in the candidate MCP tool list. 12 Role Requirements Task Instructions You are an expert bioinformatics software engineer specializing in converting command-line tools into Model Context Protocol (MCP) server tools. Your role is to analyze bioinformatics tool documentation,and make an MCP server based on that tool. You only need t...
-
[7]
Parse all available tool documentation
-
[8]
Extract all internal subcommands/tools and implement a separate Python function for each
-
[9]
Identify: * All CLI parameters (positional & optional), including Input Data, and Advanced options * Parameter types (str, int, float, bool, Path, etc.) * Default values (MUST match the parameter’s type) * Parameter constraints (e.g., value ranges, required if another is set) * Tool requirements and dependencies
-
[10]
For each internal tool/subcommand, create: * A dedicated Python function * Use the @mcp.tool() decorator with a helpful docstring
-
[11]
Parameter Handling: * For Non-optional paramater, provide a valid default (e.g., 0, 1.0, False) or use Optional[int] = None only if it is truly optional * Validate parameter values explicitly using if checks
-
[12]
File Handling: * Validate input/output file paths using Pathlib * Use tempfile if temporary files are needed * Check if files exist when necessary
-
[13]
Subprocess Execution: * Use subprocess.run(..., check=True) to execute tools * Capture and return stdout/stderr * Catch CalledProcessError and return structured error info
-
[14]
\"\"Short docstring explaining the internal tool's purpose\
Return Structured Output: * Include command_executed, stdout, stderr, and output_files (if any) Code Structure: ```python @mcp.tool() def {tool_name}( ... ): \"\"\"Short docstring explaining the internal tool's purpose\"\"\" # Input validation # File path handling # Subprocess execution # Error handling # Structured result return return { "command_execute...
-
[15]
Use explicit parameter definitions only (NEVER USE **kwargs)
-
[16]
NEVER use None as a default for non-optional int, float, or bool
-
[17]
NO NEED to import mcp
-
[18]
ALWAYS write type-safe and validated parameters
-
[19]
ONE Python function per subcommand/internal tool
-
[20]
INCLUDE helpful docstrings for every MCP tool Figure 6: Structured system prompt template for BioinfoMCP Compiler
-
[21]
Respect datatype compatibility between consecutive operations.,→
-
[22]
Prefer workflow-stage orderings that are biologically plausible.,→
-
[23]
Avoid redundant or unrelated tools
-
[24]
If multiple tools can implement the same operation, choose the most relevant,→ one based on datatype compatibility and task semantics
-
[25]
operation_path
Do not invent MCP servers or tool names. Return strict JSON only: { "operation_path": [ { "step_id": integer, "operation": string, "input_datatypes": list of strings, "output_datatypes": list of strings, "purpose": string } ], "tool_plan": [ { "step_id": integer, "operation": string, "selected_tool": string, "selected_server": string, "arguments_hint": ob...
-
[26]
Follow the operation-level workflow scaffold unless observations indicate a,→ necessary correction
-
[27]
Use only registered MCP tools and permitted local code execution.,→
-
[28]
Inspect tool outputs before proceeding to downstream steps.,→
-
[29]
If a tool call fails, diagnose the error and retry with corrected parameters,→ when appropriate
-
[30]
Maintain intermediate artifacts in the permitted run directory.,→
-
[31]
Do not access benchmark results, sibling tasks, or disallowed directories.,→
-
[32]
Produce a concise final answer and list generated result artifacts.,→ Final response format: - Summary of executed workflow - Final answer - Important output files - Any errors encountered and how they were resolved B.5 BioAgentBench LLM-Judge Prompt BioAgentBench uses an LLM-based evaluator to assess the correctness of final agent-produced arti- facts. T...
-
[33]
Input data: {input_data_path}
-
[34]
Reference data: {reference_data_path or "<none>"}
-
[35]
Processing tree: {compact_tree} Trace path mentions: {path_mentions or "<none>"}
-
[36]
Predicted result files (direct content): {result_payloads_json}
-
[37]
Ground-truth result files (direct content): {truth_payloads_json}
-
[38]
Superset containment evidence: {containment_evidence_json}
-
[39]
Prompt: {task_prompt} Task expected pipeline steps: {pipeline_steps_json} Scoring rubric (results_match from 0.000 to 1.000): - 1.0: Answer/output is fully correct and matches truth semantics and required schema.,→ - 0.7-0.9: Mostly correct with small non-critical differences (formatting/minor naming variation).,→ - 0.4-0.6: Partially correct; core direct...
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.