Non-covalent Interactions at cm⁻¹ Accuracy: Data Efficient Physics-Informed Distillation for Machine Learning Interatomic Potentials
Pith reviewed 2026-06-28 03:26 UTC · model grok-4.3
The pith
Distilling from a pretrained universal MLIP transfers physical priors on length scales and anisotropy, letting CCSD(T) fine-tuning reach cm^{-1} accuracy on non-covalent interactions with 60% less high-fidelity data.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
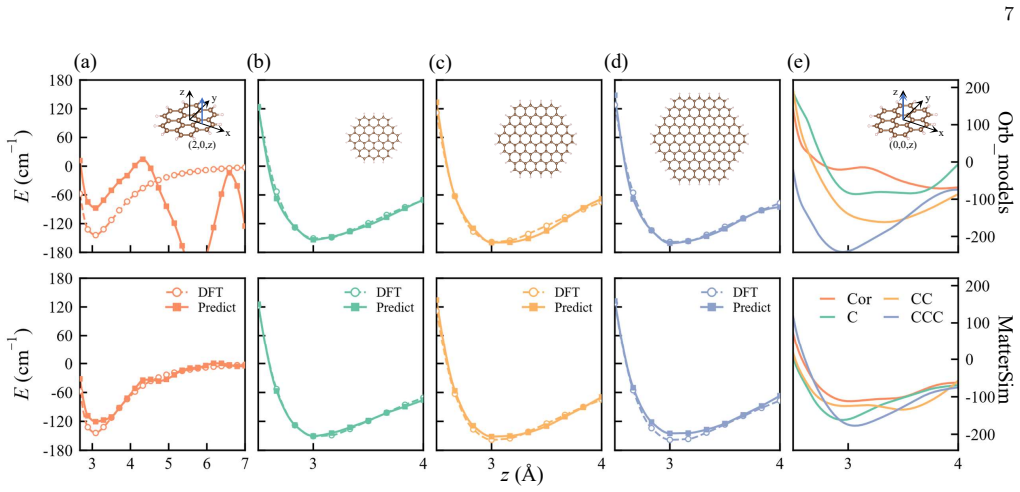
Knowledge distillation from a pretrained universal MLIP followed by CCSD(T) fine-tuning transfers a physically meaningful prior on interaction length scales, anisotropy, and repulsive-dispersive balance that CCSD(T) data then sharpens to cm^{-1} accuracy; for He-benzene this yields a 60% reduction in high-fidelity compute, and swapping the MLIP teacher changes coronene error by an order of magnitude while larger PAHs remain stable.
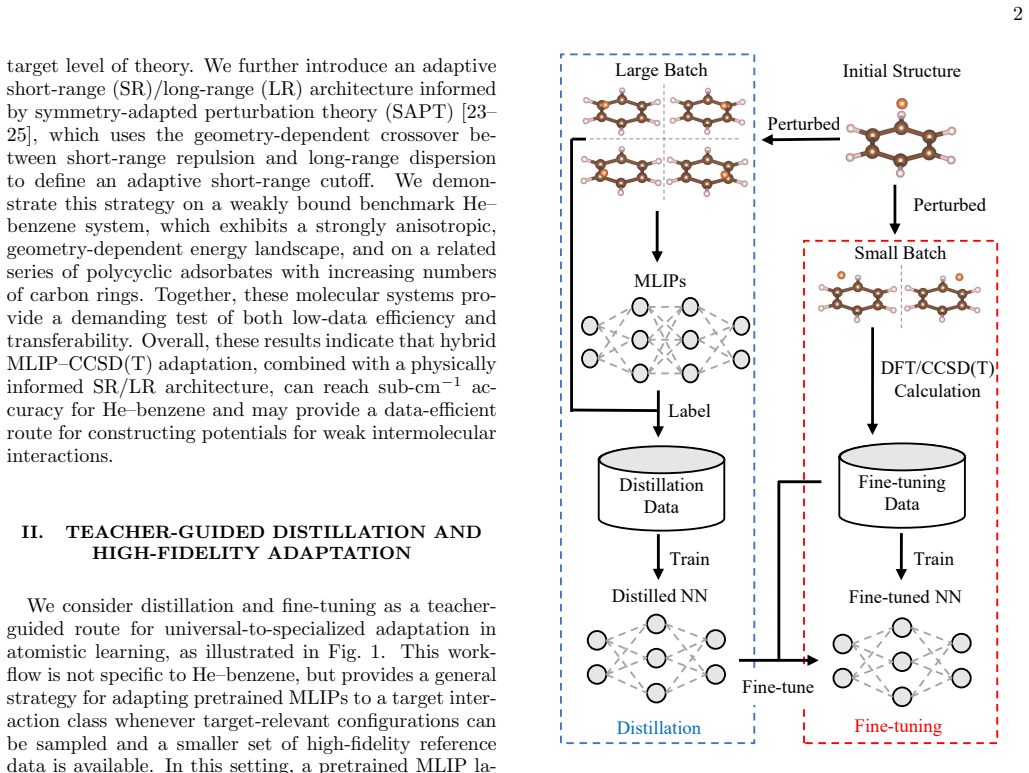
What carries the argument
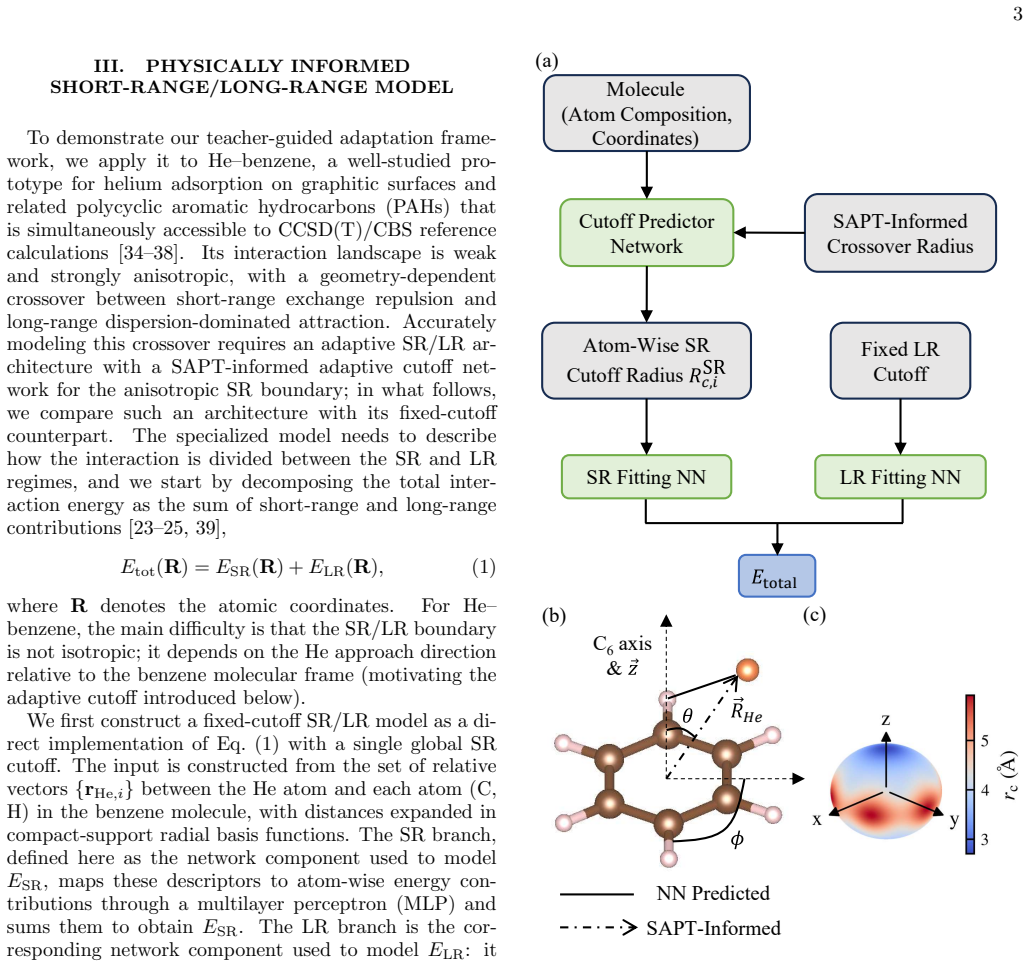
The distillation pipeline that first trains on labels from a universal MLIP teacher then fine-tunes on CCSD(T) data, augmented by an SAPT-informed adaptive short-range/long-range architecture.
If this is right
- For He-benzene, fine-tuning with 30% of the CCSD(T) data outperforms direct training on the full 80%.
- The SAPT-informed architecture lowers validation MAE from 0.75 cm^{-1} to 0.49 cm^{-1}.
- Across the circumarene PAH series, teacher choice affects accuracy on coronene far more than on larger members, indicating transferred physical structure.
- The method reduces the high-fidelity compute budget by 60% while reaching quantum-chemical accuracy.
Where Pith is reading between the lines
- The same distillation step could be tested on other classes of non-covalent interactions to check whether the length-scale and anisotropy priors remain useful.
- If the transferred prior is genuinely physical, the approach should improve data efficiency even when the target molecule lies outside the original universal MLIP training distribution.
- Replacing the universal teacher with a different foundation model would provide a direct test of how much the quality of the initial physical prior matters.
Load-bearing premise
The pretrained universal MLIP already encodes transferable physical structure on interaction length scales, anisotropy, and repulsive-dispersive balance rather than only statistical correlations that happen to fit the target molecules.
What would settle it
Running the identical pipeline with multiple different universal MLIP teachers and finding no order-of-magnitude change in coronene error, or finding that direct CCSD(T) training without distillation matches the 60% data reduction, would falsify the claim that physical structure is being transferred.
Figures

read the original abstract
Foundation models in atomistic machine learning encode interaction physics across diverse atomic environments, but whether that structure can be transferred when building specialist potentials at quantum-chemical accuracy remains open. Here we show that knowledge distillation from a pretrained universal machine-learning interatomic potential (MLIP), followed by coupled-cluster fine-tuning with single and double excitations and perturbative triples [CCSD(T)], transfers not only low-cost labels but a physically meaningful prior on interaction length scales, anisotropy, and the repulsive-dispersive balance, which CCSD(T) data then sharpens to quantum-chemical accuracy. For He--benzene, fine-tuning with 30% of the CCSD(T) data outperforms direct training using the full 80%; a 60% reduction in the high-fidelity compute budget. A symmetry-adapted perturbation theory (SAPT)-informed adaptive short-range/long-range architecture further lowers the validation MAE from 0.75 1/cm to 0.49 1/cm. Across a circumarene series of polycyclic aromatic hydrocarbons (PAHs), swapping the MLIP teacher under an otherwise identical pipeline changes the coronene error by an order of magnitude while leaving the larger PAHs stable, direct evidence that distillation transfers physical structure, not labels alone. Together, these results identify the choice of pretrained teacher as a primary design axis for data-efficient quantum-chemical-accuracy potentials, alongside architecture and training protocol.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript claims that knowledge distillation from a pretrained universal MLIP followed by CCSD(T) fine-tuning transfers not only labels but physically meaningful priors on interaction length scales, anisotropy, and repulsive-dispersive balance. This enables data-efficient quantum-chemical accuracy, with 30% CCSD(T) data outperforming full 80% direct training on He-benzene (MAE reduced further by SAPT-informed architecture to 0.49 cm^{-1}), and teacher swapping on a circumarene PAH series changing coronene error by an order of magnitude while larger systems remain stable, presented as direct evidence of physical structure transfer rather than labels alone.
Significance. If the interpretation of the teacher-swap experiment as evidence of transferred physical priors holds, the work would identify the pretrained teacher as a primary design axis for data-efficient specialist MLIPs at cm^{-1} accuracy, complementing architecture and protocol choices.
major comments (1)
- [Circumarene series results (abstract)] Circumarene series results (abstract): the claim that teacher swapping provides 'direct evidence that distillation transfers physical structure, not labels alone' is under-determined. The reported order-of-magnitude coronene error change does not isolate whether the distilled starting points differ in explicit functional forms (distance-dependent repulsion, angular dependence) versus merely supplying different basins for subsequent CCSD(T) fine-tuning; without pre-fine-tuning analysis showing the claimed physical distinctions already encoded and predictive of the final gap, the physical-prior interpretation remains at risk.
Simulated Author's Rebuttal
We thank the referee for their careful reading and the constructive comment regarding the interpretation of the circumarene series results. We address the point below.
read point-by-point responses
-
Referee: Circumarene series results (abstract): the claim that teacher swapping provides 'direct evidence that distillation transfers physical structure, not labels alone' is under-determined. The reported order-of-magnitude coronene error change does not isolate whether the distilled starting points differ in explicit functional forms (distance-dependent repulsion, angular dependence) versus merely supplying different basins for subsequent CCSD(T) fine-tuning; without pre-fine-tuning analysis showing the claimed physical distinctions already encoded and predictive of the final gap, the physical-prior interpretation remains at risk.
Authors: We agree that the current presentation of the teacher-swap experiment does not fully isolate explicit functional differences in the distilled models prior to fine-tuning. The observed order-of-magnitude difference in coronene error under identical fine-tuning data and protocol is consistent with transfer of physical structure from the teacher, but additional pre-fine-tuning diagnostics would strengthen the claim. In the revised manuscript we will add a direct comparison of the pre-fine-tuning interaction curves (radial and angular dependence) obtained from the different distilled models and relate these differences to the final accuracy gaps on coronene. We will also revise the abstract wording to indicate that the experiment provides supporting rather than conclusive evidence for physical-prior transfer. revision: partial
Circularity Check
No significant circularity; results rely on external pretrained MLIPs and independent CCSD(T) labels.
full rationale
The paper reports empirical outcomes from knowledge distillation using external universal MLIPs followed by fine-tuning on separate CCSD(T) datasets. No derivation steps, equations, or predictions reduce by construction to quantities fitted inside the same study. The teacher-swap result on coronene is an external experimental observation rather than a self-referential fit. No self-citations are invoked as load-bearing uniqueness theorems or ansatzes. The approach is self-contained against external benchmarks.
Axiom & Free-Parameter Ledger
Reference graph
Works this paper leans on
-
[1]
Hobza and J
P. Hobza and J. Rezac, Introduction: noncovalent inter- actions, Chem. Rev.116, 4911 (2016)
2016
-
[2]
M¨ uller-Dethlefs and P
K. M¨ uller-Dethlefs and P. Hobza, Noncovalent interac- tions: a challenge for experiment and theory, Chem. Rev. 100, 143 (2000)
2000
-
[3]
C. D. Sherrill, Energy component analysis ofπinterac- tions, Acc. Chem. Res.46, 1020 (2013)
2013
-
[4]
G. D. Purvis III and R. J. Bartlett, A full coupled-cluster singles and doubles model: The inclusion of disconnected triples, J. Chem. Phys.76, 1910 (1982)
1910
-
[5]
Raghavachari, G
K. Raghavachari, G. W. Trucks, J. A. Pople, and M. Head-Gordon, A fifth-order perturbation comparison of electron correlation theories, Chem. Phys. Lett.157, 479 (1989)
1989
-
[6]
Behler and M
J. Behler and M. Parrinello, Generalized neural-network representation of high-dimensional potential-energy sur- faces, Phys. Rev. Lett.98, 146401 (2007)
2007
-
[7]
M. Rupp, A. Tkatchenko, K.-R. M¨ uller, and O. A. Von Lilienfeld, Fast and accurate modeling of molecular atomization energies with machine learning, Phys. Rev. Lett.108, 058301 (2012)
2012
-
[8]
SchNet: A continuous-filter convolutional neural network for modeling quantum interactions
K. Sch¨ utt, P.-J. Kindermans, H. E. Sauceda Felix, S. Chmiela, A. Tkatchenko, and K.-R. M¨ uller, Schnet: A continuous-filter convolutional neural network for model- ing quantum interactions, Adv. Neural Inf. Process. Syst. 30, 10.48550/arXiv.1706.08566 (2017)
work page internal anchor Pith review Pith/arXiv arXiv doi:10.48550/arxiv.1706.08566 2017
-
[9]
Batzner, A
S. Batzner, A. Musaelian, L. Sun, M. Geiger, J. P. Mailoa, M. Kornbluth, N. Molinari, T. E. Smidt, and B. Kozinsky, E (3)-equivariant graph neural networks for data-efficient and accurate interatomic potentials, Nat. Commun.13, 2453 (2022)
2022
-
[10]
O. T. Unke, S. Chmiela, H. E. Sauceda, M. Gastegger, I. Poltavsky, K. T. Schutt, A. Tkatchenko, and K.-R. Muller, Machine learning force fields, Chem. Rev.121, 10142 (2021)
2021
-
[11]
Batatia, D
I. Batatia, D. P. Kovacs, G. Simm, C. Ortner, and G. Cs´ anyi, MACE: Higher order equivariant message passing neural networks for fast and accurate force fields, Adv. Neural Inf. Process. Syst.35, 11423 (2022)
2022
-
[12]
I. Batatia, P. Benner, Y. Chiang, A. M. Elena, D. P. Kov´ acs, J. Riebesell, X. R. Advincula, M. Asta, M. Avay- lon, W. J. Baldwin, and others, A foundation model for atomistic materials chemistry, J. Chem. Phys.163, 10.1063/5.0297006 (2025)
-
[13]
Chen and S
C. Chen and S. P. Ong, A universal graph deep learning interatomic potential for the periodic table, Nat. Com- put. Sci.2, 718 (2022)
2022
-
[14]
B. Deng, P. Zhong, K. Jun, J. Riebesell, K. Han, C. J. Bartel, and G. Ceder, CHGNet as a pretrained universal neural network potential for charge-informed atomistic modelling, Nat. Mach. Intell.5, 1031 (2023)
2023
-
[15]
H. Yang, C. Hu, Y. Zhou, X. Liu, Y. Shi, J. Li, G. Li, Z. Chen, S. Chen, C. Zeni, and others, Mattersim: A deep learning atomistic model across elements, temper- atures and pressures, arXiv preprint arXiv:2405.04967 10.48550/arXiv.2405.04967 (2024). 9
work page internal anchor Pith review Pith/arXiv arXiv doi:10.48550/arxiv.2405.04967 2024
-
[16]
M. Neumann, J. Gin, B. Rhodes, S. Bennett, Z. Li, H. Choubisa, A. Hussey, and J. Godwin, Orb: A fast, scalable neural network potential, arXiv preprint arXiv:2410.22570 10.48550/arXiv.2410.22570 (2024)
-
[17]
M. S. Chen, J. Lee, H.-Z. Ye, T. C. Berkelbach, D. R. Reichman, and T. E. Markland, Machine learning poten- tials from transfer learning of periodic correlated elec- tronic structure methods: Application to liquid water with AFQMC, CCSD, and CCSD (T), arXiv preprint arXiv:2211.16619 10.48550/arXiv.2211.16619 (2022)
-
[18]
J. S. Smith, B. T. Nebgen, R. Zubatyuk, N. Lubbers, C. Devereux, K. Barros, S. Tretiak, O. Isayev, and A. E. Roitberg, Approaching coupled cluster accuracy with a general-purpose neural network potential through trans- fer learning, Nat. Commun.10, 2903 (2019)
2019
-
[19]
W. Zhang, X. Wu, C. Wang, S. Hu, Y. Liu, and L.-W. Wang, Constructing machine learning interatomic poten- tials with minimum amount of ab initio data, npj Com- put. Mater. 10.1038/s41524-026-02023-y (2026)
-
[20]
R. Wang, Y. Gao, H. Wu, and Z. Zhong, Pre-training, fine-tuning, and distillation (PFD): Automatically gen- erating machine learning force fields from universal mod- els, arXiv preprint arXiv:2502.20809 10.1103/sbz6-btz8 (2025)
-
[21]
Zaverkin, D
V. Zaverkin, D. Holzm¨ uller, L. Bonfirraro, and J. K¨ astner, Transfer learning for chemically accurate in- teratomic neural network potentials, Phys. Chem. Chem. Phys.25, 5383 (2023)
2023
-
[22]
E. J. Hu, Y. Shen, P. Wallis, Z. Allen-Zhu, Y. Li, S. Wang, L. Wang, W. Chen, and others, Lora: Low-rank adaptation of large language models., Iclr1, 3 (2022)
2022
-
[23]
Jeziorski, R
B. Jeziorski, R. Moszynski, and K. Szalewicz, Perturba- tion theory approach to intermolecular potential energy surfaces of van der Waals complexes, Chem. Rev.94, 1887 (1994)
1994
-
[24]
E. G. Hohenstein and C. D. Sherrill, Wavefunction meth- ods for noncovalent interactions, Wiley Interdiscip. Rev.: Comput. Mol. Sci.2, 304 (2012)
2012
-
[25]
R. M. Parrish, L. A. Burns, D. G. Smith, A. C. Simmon- ett, A. E. DePrince III, E. G. Hohenstein, U. Bozkaya, A. Y. Sokolov, R. Di Remigio, R. M. Richard, and others, Psi4 1.1: An open-source electronic structure program emphasizing automation, advanced libraries, and inter- operability, J. Chem. Theory Comput.13, 3185 (2017)
2017
-
[26]
Distilling the Knowledge in a Neural Network
G. Hinton, O. Vinyals, and J. Dean, Distilling the knowledge in a neural network, arXiv preprint arXiv:1503.02531 10.48550/arXiv.1503.02531 (2015)
work page internal anchor Pith review Pith/arXiv arXiv doi:10.48550/arxiv.1503.02531 2015
-
[27]
Buciluˇ a, R
C. Buciluˇ a, R. Caruana, and A. Niculescu-Mizil, Model compression, inProceedings of the 12th ACM SIGKDD international conference on Knowledge discovery and data mining(2006) pp. 535–541
2006
-
[28]
J. Gou, B. Yu, S. J. Maybank, and D. Tao, Knowledge distillation: A survey, Int. J. Comput. Vis.129, 1789 (2021)
2021
-
[29]
S. J. Pan and Q. Yang, A survey on transfer learning, IEEE Trans. Knowl. Data Eng.22, 1345 (2009)
2009
-
[30]
Vandermause, S
J. Vandermause, S. B. Torrisi, S. Batzner, Y. Xie, L. Sun, A. M. Kolpak, and B. Kozinsky, On-the-fly active learn- ing of interpretable Bayesian force fields for atomistic rare events, npj Comput. Mater.6, 20 (2020)
2020
-
[31]
C. Schran, K. Brezina, and O. Marsalek, Commit- tee neural network potentials control generalization er- rors and enable active learning, J. Chem. Phys.153, 10.1063/5.0016004 (2020)
-
[32]
J. L. Gardner, K. T. Baker, and V. L. Deringer, Syn- thetic pre-training for neural-network interatomic poten- tials, Mach. Learn.: Sci. Technol.5, 015003 (2024)
2024
-
[33]
Supplementary material (2026)
2026
-
[34]
Akram, S
S. Akram, S. Paul, C. Kovacs, V. Maroulas, A. Del Mae- stro, and K. D. Vogiatzis, Accurate helium–benzene po- tential: From CCSD(T) to Gaussian process regression, J. Chem. Phys.164, 114108 (2026)
2026
-
[35]
L. Shirkov, Ab initio potentials for the ground S 0 and the first electronically excited singlet S 1 states of ben- zene–helium with application to tunneling intermolecular vibrational states, J. Phys. Chem. A128, 6132 (2024)
2024
-
[36]
Cappelletti, M
D. Cappelletti, M. Bartolomei, F. Pirani, and V. Aquilanti, Molecular beam scattering experiments on benzene- rare gas systems: Probing the potential energy surfaces for the C6H6- he,- ne, and- ar dimers, J. Phys. Chem. A106, 10764 (2002)
2002
-
[37]
Brupbacher, J
T. Brupbacher, J. Makarewicz, and A. Bauder, Inter- molecular dynamics of benzene–rare gas complexes as de- rived from microwave spectra, J. Chem. Phys.101, 9736 (1994)
1994
-
[38]
Schiller, M
A. Schiller, M. Meyer, P. Martini, F. Zappa, S. A. Kras- nokutski, F. Calvo, and P. Scheier, Adsorption of helium on small cationic PAHs: Influence of hydrocarbon struc- ture on the microsolvation pattern, J. Phys. Chem. A 125, 7813 (2021)
2021
-
[39]
Tang and J
K.-T. Tang and J. P. Toennies, An improved simple model for the van der Waals potential based on univer- sal damping functions for the dispersion coefficients, J. Chem. Phys.80, 3726 (1984)
1984
-
[40]
Y. Ji, J. Liang, and Z. Xu, Machine-learning interatomic potentials for long-range systems, Phys. Rev. Lett.135, 178001 (2025)
2025
-
[41]
A. Musaelian, S. Batzner, A. Johansson, L. Sun, C. J. Owen, M. Kornbluth, and B. Kozinsky, Learning lo- cal equivariant representations for large-scale atomistic dynamics, Nature Communications14, 10.1038/s41467- 023-36329-y (2023)
-
[42]
Zhang, A
D. Zhang, A. Peng, C. Cai, W. Li, Y. Zhou,et al., A graph neural network for the era of large atomistic mod- els, npj Computational Materials (2026)
2026
-
[43]
Batzner, A
S. Batzner, A. Musaelian, L. Sun, M. Geiger, J. P. Mailoa, M. Kornbluth, N. Molinari, T. E. Smidt, and B. Kozinsky, E(3)-equivariant graph neural networks for data-efficient and accurate interatomic potentials, Nature Communications13, 2453 (2022)
2022
-
[44]
B. Deng, P. Zhong, K. Jun, J. Riebesell, K. Han, C. J. Bartel, and G. Ceder, Chgnet as a pretrained universal neural network potential for charge-informed atomistic modelling, Nature Machine Intelligence5, 1031–1041 (2023)
2023
-
[45]
X. Fu, B. M. Wood, L. Barroso-Luque, D. S. Levine, M. Gao, M. Dzamba, and C. L. Zitnick, Learning smooth and expressive interatomic potentials for physical prop- erty prediction, inProceedings of the 42nd International Conference on Machine Learning, Proceedings of Ma- chine Learning Research, Vol. 267, edited by A. Singh, M. Fazel, D. Hsu, S. Lacoste-Jul...
2025
-
[46]
B. M. Wood, M. Dzamba, X. Fu, M. Gao, M. Shuaibi, L. Barroso-Luque, K. Abdelmaqsoud, V. Gharakhanyan, J. R. Kitchin, D. S. Levine, K. Michel, A. Sriram, T. Co- hen, A. Das, A. Rizvi, S. J. Sahoo, Z. W. Ulissi, and 10 C. L. Zitnick, Uma: A family of universal models for atoms (2026), arXiv:2506.23971 [cs.LG]
arXiv 2026
-
[47]
Y. Zhou, S. Hu, X. Zhang, H. Wang, G. Tan, and W. Jia, Matris: Toward reliable and efficient pretrained machine learning interatomic potentials (2026), arXiv:2603.02002 [cs.LG]
arXiv 2026
-
[48]
F. Bigi, P. Pegolo, A. Mazitov, J. Schmidt, and M. Ce- riotti, Pushing the limits of unconstrained machine- learned interatomic potentials (2026), arXiv:2601.16195 [physics.chem-ph]
Pith/arXiv arXiv 2026
-
[49]
B. Yin, J. Wang, W. Du, P. Wang, P. Ying, H. Jia, Z. Zhang, Y. Du, C. Gomes, C. Duan, G. Henkelman, and H. Xiao, Alphanet: scaling up local-frame-based neural network interatomic potentials, npj Computational Ma- terials11, 10.1038/s41524-025-01817-w (2025)
-
[50]
J. Kim, J. Kim, J. Kim, J. Lee, Y. Park, Y. Kang, and S. Han, Data-efficient multifidelity training for high- fidelity machine learning interatomic potentials, J. Am. Chem. Soc.147, 1042 (2024)
2024
-
[51]
T. Li, W. Li, A. Peng, J. Xue, L. Zhang, D. Zhang, and H. Wang, Dpa4: Pushing the accuracy-cost frontier of in- teratomic potentials with emfa so(2) convolution (2026), arXiv:2606.02419 [physics.chem-ph]
Pith/arXiv arXiv 2026
-
[52]
K. Yan, M. Bohde, A. Kryvenko, Z. Xiang, K. Zhao, S. Zhu, S. Kolachina, D. Sarıt¨ urk, J. Xie, R. Arroy- ave, X. Qian, X. Qian, and S. Ji, A materials founda- tion model via hybrid invariant-equivariant architectures (2025), arXiv:2503.05771 [cs.LG]
arXiv 2025
-
[53]
Y. Lysogorskiy, A. Bochkarev, and R. Drautz, Graph atomic cluster expansion for foundational machine learn- ing interatomic potentials, npj Computational Materials 12, 10.1038/s41524-026-01979-1 (2026)
-
[54]
Y.-L. Liao, B. Wood, A. Das*, and T. Smidt*, EquiformerV2: Improved Equivariant Transformer for Scaling to Higher-Degree Representations, inInterna- tional Conference on Learning Representations (ICLR) (2024)
2024
-
[55]
Burke, Perspective on density functional theory, J
K. Burke, Perspective on density functional theory, J. Chem. Phys.136, 10.1063/1.4704546 (2012)
-
[56]
Lazar, F
P. Lazar, F. Karlicky, P. Jurecka, M. Kocman, E. Otyep- kov´ a, K. Safarova, and M. Otyepka, Adsorption of small organic molecules on graphene, J. Am. Chem. Soc.135, 6372 (2013)
2013
-
[57]
Zaspel, B
P. Zaspel, B. Huang, H. Harbrecht, and O. A. Von Lilien- feld, Boosting quantum machine learning models with a multilevel combination technique: Pople diagrams revis- ited, J. Chem. Theory Comput.15, 1546 (2018)
2018
-
[58]
Ramakrishnan, P
R. Ramakrishnan, P. O. Dral, M. Rupp, and O. A. Von Lilienfeld, Big data meets quantum chemistry ap- proximations: the ∆-machine learning approach, J. Chem. Theory Comput.11, 2087 (2015)
2087
-
[59]
J. S. Smith, R. Zubatyuk, B. Nebgen, N. Lubbers, K. Barros, A. E. Roitberg, O. Isayev, and S. Tretiak, The ANI-1ccx and ANI-1x data sets, coupled-cluster and den- sity functional theory properties for molecules, Sci. Data 7, 134 (2020)
2020
-
[60]
Zhang, D.-Y
L. Zhang, D.-Y. Lin, H. Wang, R. Car, and W. E, Active learning of uniformly accurate interatomic potentials for materials simulation, Phys. Rev. Mater.3, 023804 (2019)
2019
-
[61]
E. Uteva, R. S. Graham, R. D. Wilkinson, and R. J. Wheatley, Active learning in Gaussian process interpo- lation of potential energy surfaces, J. Chem. Phys.149, 10.1063/1.5051772 (2018)
-
[62]
Non-covalent Interactions at cm −1 Accuracy: Data Efficient Physics-Informed Distillation for Machine Learning Interatomic Potentials
Y. Shen and A. Del Maestro, MLIP distillation and fine-tuning for He–benzene and PAH weak interactions: GitHub Repository (2026). Supplemental Material for “Non-covalent Interactions at cm −1 Accuracy: Data Efficient Physics-Informed Distillation for Machine Learning Interatomic Potentials” Yulin Shen,1 Shahzad Akram, 2 Louis Primeau, 1 Gen Zu, 3 Konstant...
2026
-
[63]
Akram, S
S. Akram, S. Paul, C. Kovacs, V. Maroulas, A. Del Maestro, and K. D. Vogiatzis, Accurate helium–benzene potential: From CCSD(T) to Gaussian process regression, J. Chem. Phys.164, 114108 (2026)
2026
-
[64]
H. Wang, L. Zhang, J. Han, and others, DeePMD-kit: A deep learning package for many-body potential energy represen- tation and molecular dynamics, Comput. Phys. Commun.228, 178 (2018)
2018
-
[65]
J. Zeng, D. Zhang, D. Lu, P. Mo, Z. Li, Y. Chen, M. Rynik, L. Huang, Z. Li, S. Shi, Y. Wang, H. Ye, P. Tuo, J. Yang, Y. Ding, Y. Li, D. Tisi, Q. Zeng, H. Bao, Y. Xia, J. Huang, K. Muraoka, Y. Wang, J. Chang, F. Yuan, S. L. Bore, C. Cai, Y. Lin, B. Wang, J. Xu, J.-X. Zhu, C. Luo, Y. Zhang, R. E. A. Goodall, W. Liang, A. K. Singh, S. Yao, J. Zhang, R. Wentz...
2023
-
[66]
J. Zeng, D. Zhang, A. Peng, X. Zhang, S. He, Y. Wang, X. Liu, H. Bi, Y. Li, C. Cai, and others, DeePMD-kit v3: a multiple-backend framework for machine learning potentials, J. Chem. Theory Comput.21, 4375 (2025). 15
2025
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.