p-adic Bi-Filtrations for Topological Machine Learning on Genomic Sequences
Pith reviewed 2026-06-27 22:44 UTC · model grok-4.3
The pith
pVR encodes genomic sequences via a bi-filtration of p-adic prefix distances and k-mer frequency distances to extract topological features for classification.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
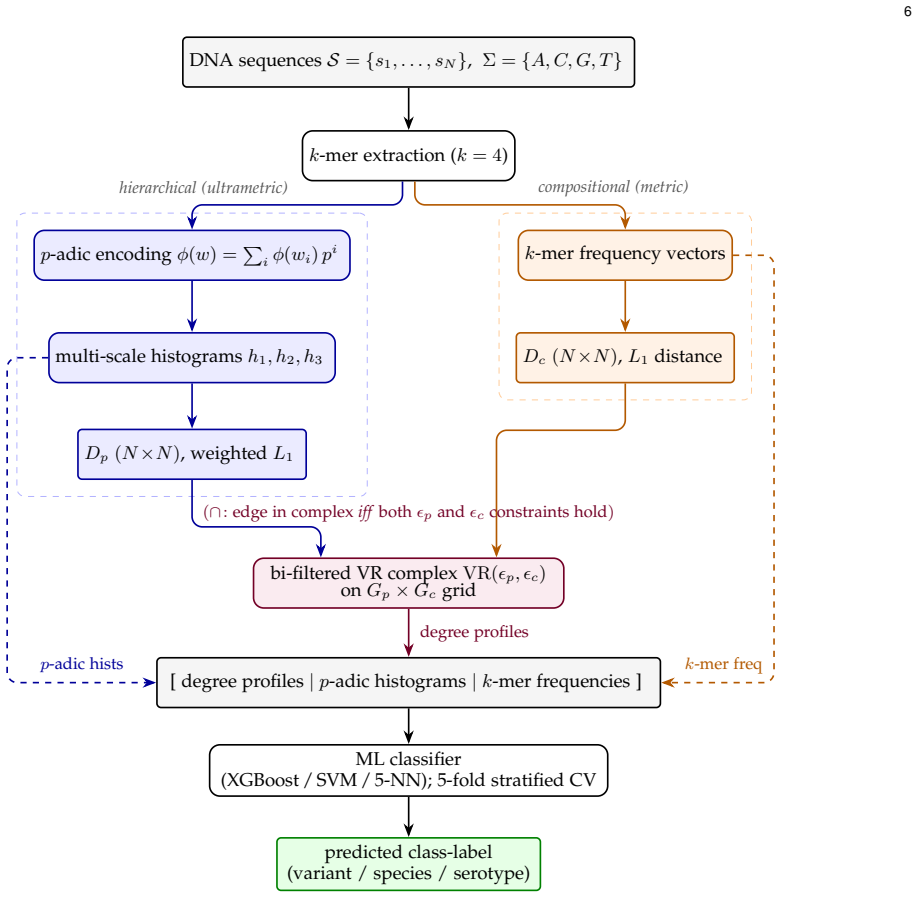
pVR jointly parameterizes a bi-filtered Vietoris-Rips complex by a p-adic metric on k-mer prefixes and an L1 metric on k-mer frequencies; the persistent homology of this bi-filtration yields stable, prime-invariant topological summaries that serve as features improving classification accuracy on low-sample genomic datasets relative to alignment-free baselines.
What carries the argument
bi-filtered Vietoris-Rips complex jointly parameterized by p-adic distance on k-mer prefixes and L1 distance on k-mer frequencies
If this is right
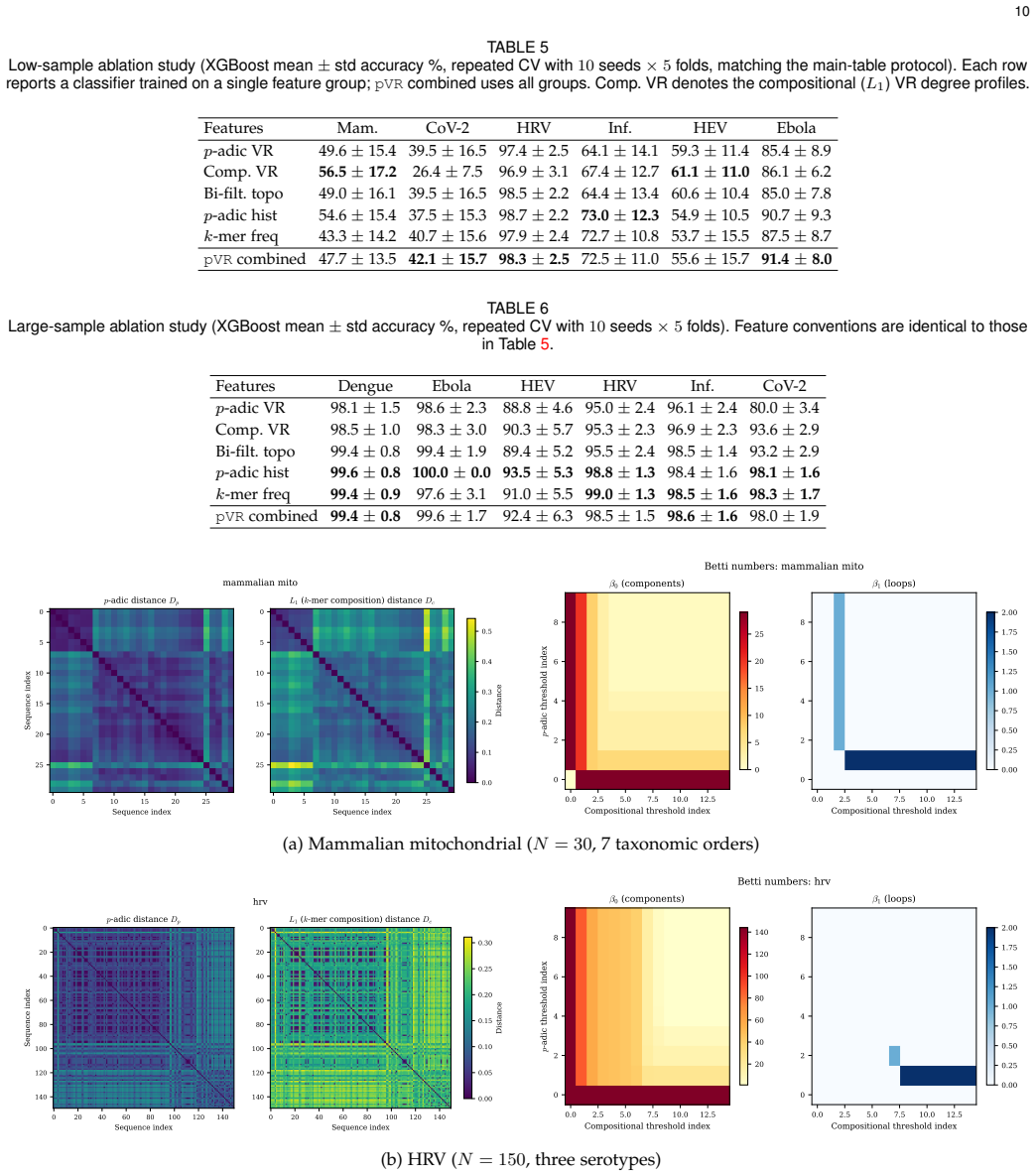
- Outperforms four alignment-free baselines on three of six low-sample datasets by as much as 21 percentage points.
- Outperforms zero-shot frozen embeddings from the 500M-parameter Nucleotide Transformer v2 by 6.7 to 11.4 percentage points on three low-sample benchmarks.
- The topological summaries remain stable under metric perturbations and invariant to the choice of prime p.
- A single p-adic axis produces uninformative homology while the bi-filtration recovers nontrivial features.
Where Pith is reading between the lines
- The same bi-filtration construction could be tested on other ordered sequential data such as protein sequences or parsed text where prefix hierarchies are present.
- Accuracy may remain limited on problems with high rates of isolated substitutions, suggesting a need for hybrid filtrations that also capture pointwise differences.
- In regimes where all methods saturate, the topological features might still reduce variance when combined with sequence kernels or learned embeddings.
Load-bearing premise
The p-adic distance on k-mer prefixes captures meaningful hierarchical positional structure in the genomic sequences.
What would settle it
Performance gains disappear or reverse on additional genomic datasets dominated by point-mutation divergence rather than hierarchical prefix structure, such as further SARS-CoV-2 variant collections.
Figures



read the original abstract
We introduce pVR, a topological machine learning framework for alignment-free genomic sequence classification that combines $p$-adic numbers with topological data analysis. Each DNA sequence is encoded along two complementary axes: a $p$-adic distance on $k$-mer prefixes, which captures hierarchical positional structure, and a compositional $L_1$ distance on $k$-mer frequencies, which captures local sequence content. The two distances jointly parameterise a bi-filtered Vietoris--Rips complex, and per-sequence topological summaries from this bi-filtration serve as features for standard machine learning classifiers. We establish theoretical guarantees for the construction: stability under metric perturbations and invariance to the choice of prime, alongside a result that explains why a single $p$-adic axis is topologically uninformative and why the bi-filtration recovers nontrivial homology. On twelve genomic benchmarks ($28$ to $500$ sequences, $3$ to $7$ classes), pVR outperforms four established alignment-free baselines on three of six low-sample datasets, with gains of up to $21$ percentage points; it underperforms only on a SARS-CoV-2 variant benchmark whose point-mutation divergence violates the hierarchical assumption, and all methods saturate in the large-sample regime. pVR also outperforms zero-shot frozen embeddings from the 500M-parameter Nucleotide Transformer v2 by $6.7$ to $11.4$ percentage points on three low-sample benchmarks. The pVR codebase is publicly available at https://github.com/MAHI-Group/pVR.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The paper introduces pVR, a topological machine learning framework for alignment-free genomic sequence classification. It encodes DNA sequences using a p-adic distance on k-mer prefixes (capturing hierarchical positional structure) and an L1 distance on k-mer frequencies (capturing compositional content), jointly parameterizing a bi-filtered Vietoris-Rips complex whose topological summaries serve as features for standard classifiers. Theoretical guarantees are claimed for stability under metric perturbations, invariance to prime choice, and the necessity of the bi-filtration (single p-adic axis being topologically trivial). On twelve genomic benchmarks (28-500 sequences, 3-7 classes), pVR outperforms four alignment-free baselines on three of six low-sample datasets (gains up to 21 pp), underperforms on a SARS-CoV-2 variant set violating the hierarchical assumption, saturates with larger samples, and beats zero-shot 500M-parameter Nucleotide Transformer embeddings by 6.7-11.4 pp on three low-sample tasks. Code is publicly available.
Significance. If the results hold, the work offers a novel integration of p-adic valuations into TDA for genomics that exploits hierarchical structure in sequences, with demonstrated utility in low-sample regimes where it surpasses both classical alignment-free methods and large frozen embeddings. The public codebase is a clear strength, enabling direct verification of the bi-filtration construction and empirical claims.
minor comments (3)
- The abstract and results section should explicitly reference the specific theorems or propositions establishing stability, prime invariance, and single-axis triviality (currently only asserted).
- Table or figure reporting the per-benchmark accuracies should include standard deviations or statistical significance tests to support the 'up to 21 percentage points' gains.
- The k-mer length k and filtration parameters are listed as free parameters; a brief sensitivity analysis or default selection protocol would strengthen the reproducibility claim.
Simulated Author's Rebuttal
We thank the referee for the detailed summary of our work and the positive evaluation of its significance, particularly regarding the integration of p-adic valuations with TDA for low-sample genomic classification and the public codebase. The recommendation for minor revision is noted. No major comments were provided in the report, so we have no points requiring point-by-point rebuttal.
Circularity Check
No significant circularity
full rationale
The paper defines a new p-adic distance on k-mer prefixes and an L1 distance on frequencies, jointly parameterizing a bi-filtered Vietoris-Rips complex whose topological summaries are used as features for off-the-shelf classifiers. Theoretical claims (stability, prime invariance, single-axis triviality) are stated as results of the construction itself rather than fitted or renamed inputs. No equations reduce a claimed prediction to a parameter fit by construction, and no load-bearing premise rests on self-citation chains. Public code supplies an independent verification path. The central claim therefore remains self-contained against external benchmarks.
Axiom & Free-Parameter Ledger
free parameters (2)
- k-mer length k
- filtration parameters
axioms (2)
- standard math p-adic numbers form a metric space suitable for hierarchical structures
- standard math Vietoris-Rips complexes are stable under small perturbations of the metric
invented entities (1)
-
p-adic bi-filtration
no independent evidence
Reference graph
Works this paper leans on
-
[1]
Alignment-free sequence comparison: benefits, applications, and tools,
A. Zielezinski, S. Vinga, J. Almeida, and W. M. Karlowski, “Alignment-free sequence comparison: benefits, applications, and tools,”Genome biology, vol. 18, no. 1, p. 186, 2017
2017
-
[2]
Alignment- free genome comparison with feature frequency profiles (ffp) and optimal resolutions,
G. E. Sims, S.-R. Jun, G. A. Wu, and S.-H. Kim, “Alignment- free genome comparison with feature frequency profiles (ffp) and optimal resolutions,”Proceedings of the National Academy of Sciences, vol. 106, no. 8, pp. 2677–2682, 2009
2009
-
[3]
A novel method of characterizing genetic sequences: genome space with biological distance and applications,
M. Deng, C. Yu, Q. Liang, R. L. He, and S. S.-T. Yau, “A novel method of characterizing genetic sequences: genome space with biological distance and applications,”PloS one, vol. 6, no. 3, p. e17293, 2011. 12 TABLE 9 Sensitivity tok-mer size: XGBoost accuracy (%, mean±std over a single 5-fold split). The primepis invariant by Proposition 12. kLow-sample da...
2011
-
[4]
Mash: fast genome and metagenome distance estimation using minhash,
B. D. Ondov, T. J. Treangen, P . Melsted, A. B. Mallonee, N. H. Bergmanet al., “Mash: fast genome and metagenome distance estimation using minhash,”Genome biology, vol. 17, no. 1, p. 132, 2016
2016
-
[5]
Topology of viral evolution,
J. M. Chan, G. Carlsson, and R. Rabadan, “Topology of viral evolution,”Proceedings of the National Academy of Sciences, vol. 110, no. 46, pp. 18 566–18 571, 2013
2013
-
[6]
Revealing the shape of genome space via k-mer topology,
Y. Hozumi and G.-W. Wei, “Revealing the shape of genome space via k-mer topology,”arXiv preprint arXiv:2412.20202, 2024
arXiv 2024
-
[7]
Cakl: Commutative algebra k-mer learning of genomics,
F. Suwayyid, Y. Hozumi, H. Feng, M. Zia, J. Wee, and G.-W. Wei, “Cakl: Commutative algebra k-mer learning of genomics,”arXiv preprint arXiv:2508.09406, 2025
arXiv 2025
-
[8]
p-adic modelling of the genome and the genetic code,
B. Dragovich and A. Dragovich, “p-adic modelling of the genome and the genetic code,”The Computer Journal, vol. 53, no. 4, pp. 432–442, 2010
2010
-
[9]
p-adic mathematics and theoretical biology,
B. Dragovich, A. Y. Khrennikov, S. V . Kozyrev, and N. ˇZ. Mi ˇsi´c, “p-adic mathematics and theoretical biology,”Biosystems, vol. 199, p. 104288, 2021
2021
-
[10]
Characterization, stability and convergence of hierarchical clustering methods
G. E. Carlsson, F. M ´emoliet al., “Characterization, stability and convergence of hierarchical clustering methods.”J. Mach. Learn. Res., vol. 11, no. 47, pp. 1425–1470, 2010
2010
-
[11]
Semple, M
C. Semple, M. Steelet al.,Phylogenetics. Oxford University Press on Demand, 2003, vol. 24
2003
-
[12]
DNABERT: pre-trained bidirectional encoder representations from transformers model for dna-language in genome,
Y. Ji, Z. Zhou, H. Liu, and R. V . Davuluri, “DNABERT: pre-trained bidirectional encoder representations from transformers model for dna-language in genome,”Bioinformatics, vol. 37, no. 15, pp. 2112– 2120, 2021
2021
-
[13]
Nucleotide transformer: building and evaluating robust foundation models for human genomics,
H. Dalla-Torre, L. Gonzalez, J. Mendoza-Revilla, N. Lopez Car- ranzaet al., “Nucleotide transformer: building and evaluating robust foundation models for human genomics,”Nature Methods, vol. 22, no. 2, pp. 287–297, 2025
2025
-
[14]
Dna language model grover learns sequence context in the human genome,
M. Sanabria, J. Hirsch, P . M. Joubert, and A. R. Poetsch, “Dna language model grover learns sequence context in the human genome,”Nature Machine Intelligence, vol. 6, no. 8, pp. 911–923, 2024
2024
-
[15]
Topological methods for genomics: present and future directions,
P . G. C ´amara, “Topological methods for genomics: present and future directions,”Current opinion in systems biology, vol. 1, pp. 95–101, 2017
2017
-
[16]
Rabadan and A
R. Rabadan and A. J. Blumberg,Topological data analysis for genomics and evolution: topology in biology. Cambridge University Press, 2019
2019
-
[17]
Ultrametrics in the genetic code and the genome,
B. Dragovich, A. Y. Khrennikov, and N. ˇZ. Mi ˇsi´c, “Ultrametrics in the genetic code and the genome,”Applied Mathematics and Computation, vol. 309, pp. 350–358, 2017
2017
-
[18]
p-clustval: a novel p-adic approach for enhanced clustering of high- dimensional single-cell rnaseq data,
P . Sharma, S. Mishra, H. Kurban, and M. Dalkilic, “p-clustval: a novel p-adic approach for enhanced clustering of high- dimensional single-cell rnaseq data,”International Journal of Data Science and Analytics, vol. 20, no. 4, pp. 4051–4066, 2025
2025
-
[19]
v-punns: van der put neural networks for transparent ultrametric representation learning,
G. L. R. N’guessan, “v-punns: van der put neural networks for transparent ultrametric representation learning,”arXiv preprint arXiv:2508.01010, 2025
arXiv 2025
-
[20]
A. F. Martins, “Learning with thep-adics,”arXiv preprint arXiv:2512.22692, 2025
arXiv 2025
-
[21]
Barcodes: the persistent topology of data,
R. Ghrist, “Barcodes: the persistent topology of data,”Bulletin of the American Mathematical Society, vol. 45, no. 1, pp. 61–75, 2008
2008
-
[22]
Topological persistence and simplification,
H. Edelsbrunner, D. Letscher, and A. Zomorodian, “Topological persistence and simplification,”Discrete & computational geometry, vol. 28, no. 4, pp. 511–533, 2002
2002
-
[23]
Computing persistent homol- ogy,
A. Zomorodian and G. Carlsson, “Computing persistent homol- ogy,” inProceedings of the twentieth annual symposium on Computa- tional geometry, 2004, pp. 347–356
2004
-
[24]
The theory of multidimensional persistence,
G. Carlsson and A. Zomorodian, “The theory of multidimensional persistence,” inProceedings of the twenty-third annual symposium on Computational geometry, 2007, pp. 184–193
2007
-
[25]
The theory of the interleaving distance on multi- dimensional persistence modules,
M. Lesnick, “The theory of the interleaving distance on multi- dimensional persistence modules,”Foundations of Computational Mathematics, vol. 15, no. 3, pp. 613–650, 2015
2015
-
[26]
XGBoost: A scalable tree boosting system,
T. Chen and C. Guestrin, “XGBoost: A scalable tree boosting system,” inProceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, 2016, pp. 785– 794
2016
-
[27]
The gudhi library: Simplicial complexes and persistent homology,
C. Maria, J.-D. Boissonnat, M. Glisse, and M. Yvinec, “The gudhi library: Simplicial complexes and persistent homology,” inInter- national congress on mathematical software. Springer, 2014, pp. 167– 174
2014
-
[28]
Inference for the generalization error,
C. Nadeau and Y. Bengio, “Inference for the generalization error,” Machine Learning, vol. 52, no. 3, pp. 239–281, Sep 2003. [Online]. Available: https://doi.org/10.1023/A:1024068626366
-
[29]
Evaluating the replicability of significance tests for comparing learning algorithms,
R. R. Bouckaert and E. Frank, “Evaluating the replicability of significance tests for comparing learning algorithms,” inPacific- Asia conference on knowledge discovery and data mining. Springer, 2004, pp. 3–12
2004
-
[30]
T. M. Mitchell,Machine Learning. McGraw-Hill, 1997
1997
-
[31]
A model of inductive bias learning,
J. Baxter, “A model of inductive bias learning,”Journal of artificial intelligence research, vol. 12, pp. 149–198, 2000
2000
-
[32]
K. Kontolati, R. J. Gladstone, I. Davis, and E. Pickering, “Biology- informed neural networks learn nonlinear representations from omics data to improve genomic prediction and interpretability,” arXiv preprint arXiv:2510.14970, 2025
arXiv 2025
-
[33]
A review of some techniques for inclusion of domain-knowledge into deep neural networks,
T. Dash, S. Chitlangia, A. Ahuja, and A. Srinivasan, “A review of some techniques for inclusion of domain-knowledge into deep neural networks,”Scientific Reports, vol. 12, no. 1, p. 1040, 2022
2022
-
[34]
T. Dash, “BIRDNet: Mining and encoding boolean implication knowledge graphs as interpretable deep neural networks,”arXiv preprint arXiv:2605.28739, 2026
Pith/arXiv arXiv 2026
-
[35]
Consensus proposals for classification of the family hepeviridae,
D. B. Smith, P . Simmonds, I. C. on the Taxonomy of Viruses Hepe- viridae Study Groupet al., “Consensus proposals for classification of the family hepeviridae,”Journal of General Virology, vol. 95, no. 10, pp. 2223–2232, 2014
2014
-
[36]
Topological estimation using witness complexes
V . De Silva and G. E. Carlsson, “Topological estimation using witness complexes.” inPBG, 2004, pp. 157–166
2004
-
[37]
Persistence images: A stable vector representation of persistent homology,
H. Adams, T. Emerson, M. Kirby, R. Neville, C. Petersonet al., “Persistence images: A stable vector representation of persistent homology,”Journal of Machine Learning Research, vol. 18, no. 8, pp. 1–35, 2017
2017
-
[38]
Learning representa- tions of persistence barcodes,
C. D. Hofer, R. Kwitt, and M. Niethammer, “Learning representa- tions of persistence barcodes,”Journal of Machine Learning Research, vol. 20, no. 126, pp. 1–45, 2019
2019
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.