RLEASE: Reinforcement Learning Efficient Active Space Engine
Pith reviewed 2026-06-27 20:02 UTC · model grok-4.3
The pith
A neural network trained by reinforcement learning selects compact active spaces that transfer to chemically diverse molecules without retraining.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
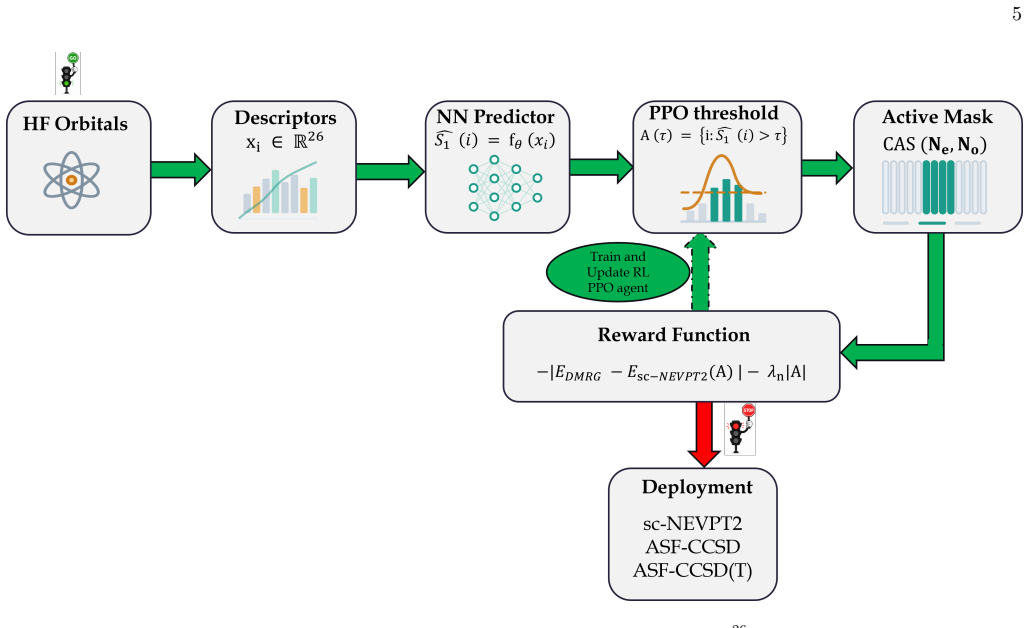
RLEASE trains a neural network to predict per-orbital scores from Hartree-Fock descriptors and optimizes a learned threshold via proximal policy optimization; the reward is the sc-NEVPT2 versus DMRG energy discrepancy on the selected active space. The resulting policy, trained on a small collection of molecules and geometries, produces compact active spaces that deliver potential-energy surfaces competitive with entropy-based selectors when applied to chemically diverse test systems, all at the cost of a single inexpensive orbital-descriptor evaluation and neural-network inference.
What carries the argument
A neural network that maps inexpensive Hartree-Fock orbital descriptors to per-orbital diagnostic scores, combined with a learned threshold policy optimized by proximal policy optimization using sc-NEVPT2-DMRG discrepancy as the reward signal.
If this is right
- High-throughput multireference calculations become possible because deployment requires only cheap orbital descriptors and a single neural-network inference.
- The same trained network can be used with multireference perturbation theory or composite coupled-cluster estimators on new molecules.
- Active-space selection no longer requires molecule-specific pilot DMRG calculations or retraining.
- Compact active spaces are produced that remain competitive with entropy-based methods across diverse chemical systems.
Where Pith is reading between the lines
- Similar reinforcement-learning policies could be explored for other selection tasks in quantum chemistry that currently rely on expensive reference calculations.
- The approach might scale to larger molecules where full DMRG references become prohibitive, provided the orbital-descriptor representation remains informative.
- Integration with existing multireference workflows could reduce the expert time currently spent on trial-and-error active-space trials.
Load-bearing premise
The reward signal derived from sc-NEVPT2 versus DMRG discrepancy on a small training set will continue to produce accurate and compact active-space choices when the same network is applied to new, chemically diverse molecules.
What would settle it
On a chemically diverse molecule outside the training distribution, the RLEASE-selected active space produces a potential-energy surface whose error relative to DMRG reference values is substantially larger than the error obtained with an established entropy-based active-space selector.
Figures
read the original abstract
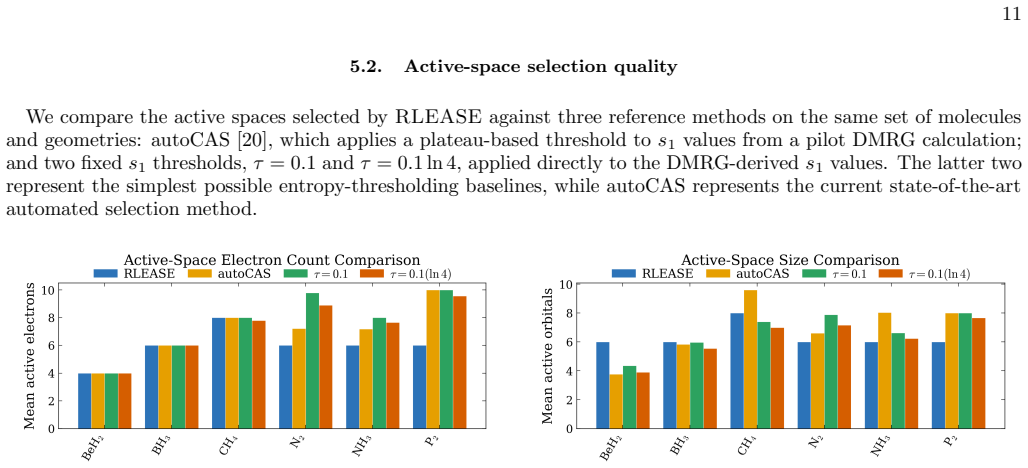
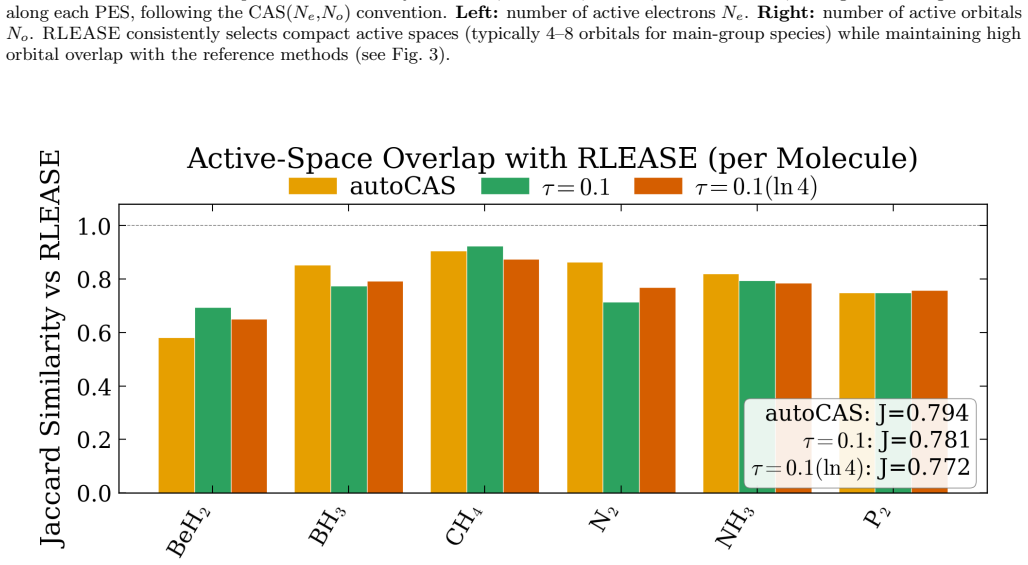
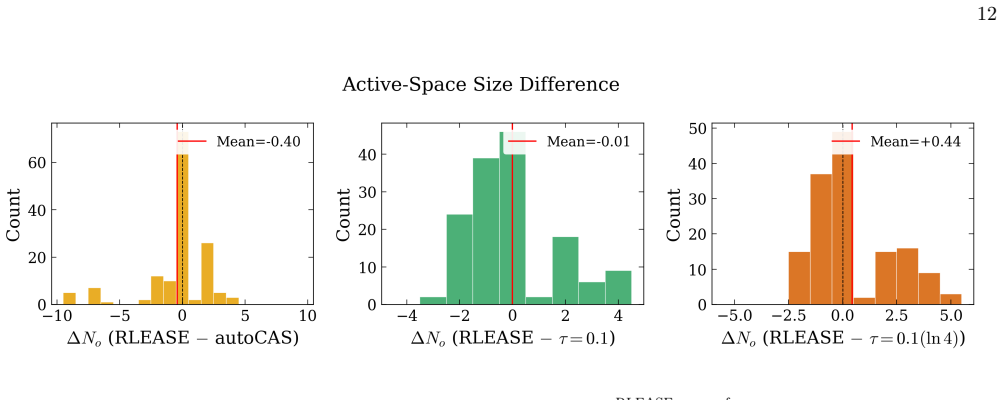
Selecting the active space for multireference electronic-structure calculations is a long-standing bottleneck that often requires expert chemical intuition and costly trial-and-error. We introduce RLEASE (Reinforcement Learning Efficient Active Space Engine), a low-cost method for automatic, geometry-dependent active-space selection. A neural network predicts per-orbital diagnostic scores ($\hat{s}_{1}$) from inexpensive Hartree-Fock orbital descriptors, and a learned threshold partitions orbitals into active and inactive sets. The threshold policy is optimized with proximal policy optimization, using the discrepancy between sc-NEVPT2 energies computed with the selected active space and DMRG reference energies as the reward. After training, the same RLEASE-selected active spaces can be used with multireference perturbation theory or composite coupled-cluster energy estimators. Despite being trained on a small set of molecules and geometries, RLEASE transfers to chemically diverse test systems, producing compact active spaces and competitive potential-energy surfaces relative to established entropy-based selectors. Because deployment requires only inexpensive orbital descriptors and neural-network inference, RLEASE enables high-throughput multireference workflows without molecule-specific retraining or target-system pilot DMRG calculations.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The paper introduces RLEASE, a reinforcement learning method (using PPO) for automatic, geometry-dependent active-space selection in multireference calculations. A neural network predicts per-orbital diagnostic scores from inexpensive Hartree-Fock orbital descriptors; a learned threshold partitions orbitals into active/inactive sets. The policy is optimized with a reward equal to the discrepancy between sc-NEVPT2 energies (using the selected space) and DMRG reference energies. After training on a small set of molecules and geometries, the fixed network is claimed to transfer to chemically diverse test systems, yielding compact active spaces whose subsequent multireference energies are competitive with those from established entropy-based selectors, all at the cost of only HF descriptors plus NN inference.
Significance. If the transferability claim holds, RLEASE would remove a major practical bottleneck for high-throughput multireference workflows by eliminating molecule-specific DMRG pilot calculations and expert trial-and-error while still producing usable active spaces. The low inference cost is a genuine practical strength.
major comments (2)
- [Abstract] Abstract: the central transferability claim ("Despite being trained on a small set of molecules and geometries, RLEASE transfers to chemically diverse test systems") is load-bearing for the deployment narrative, yet the reward is defined directly as sc-NEVPT2–DMRG discrepancy; this creates an explicit dependence on the external DMRG benchmark during training that must be shown not to produce overfitting to the training distribution's electronic motifs.
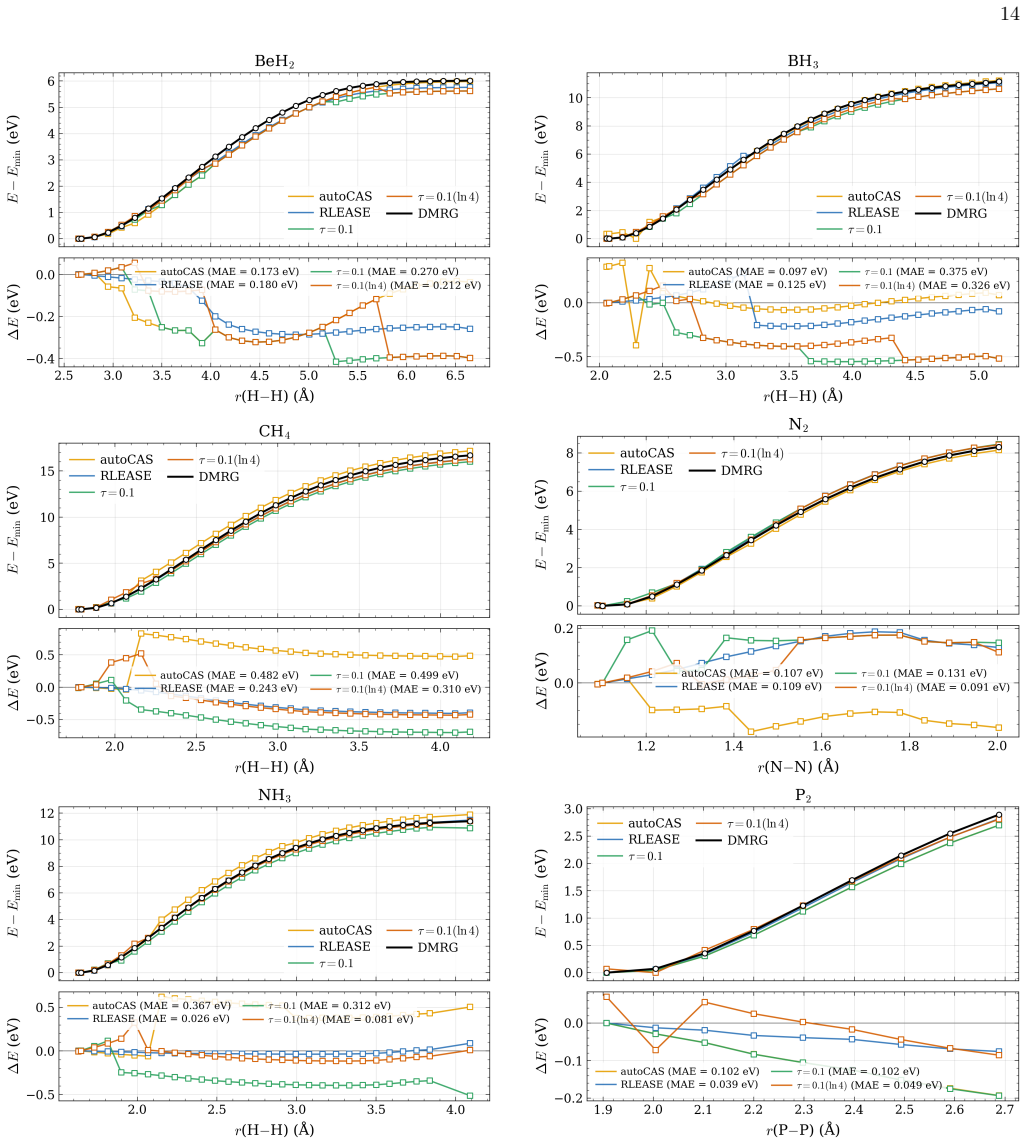
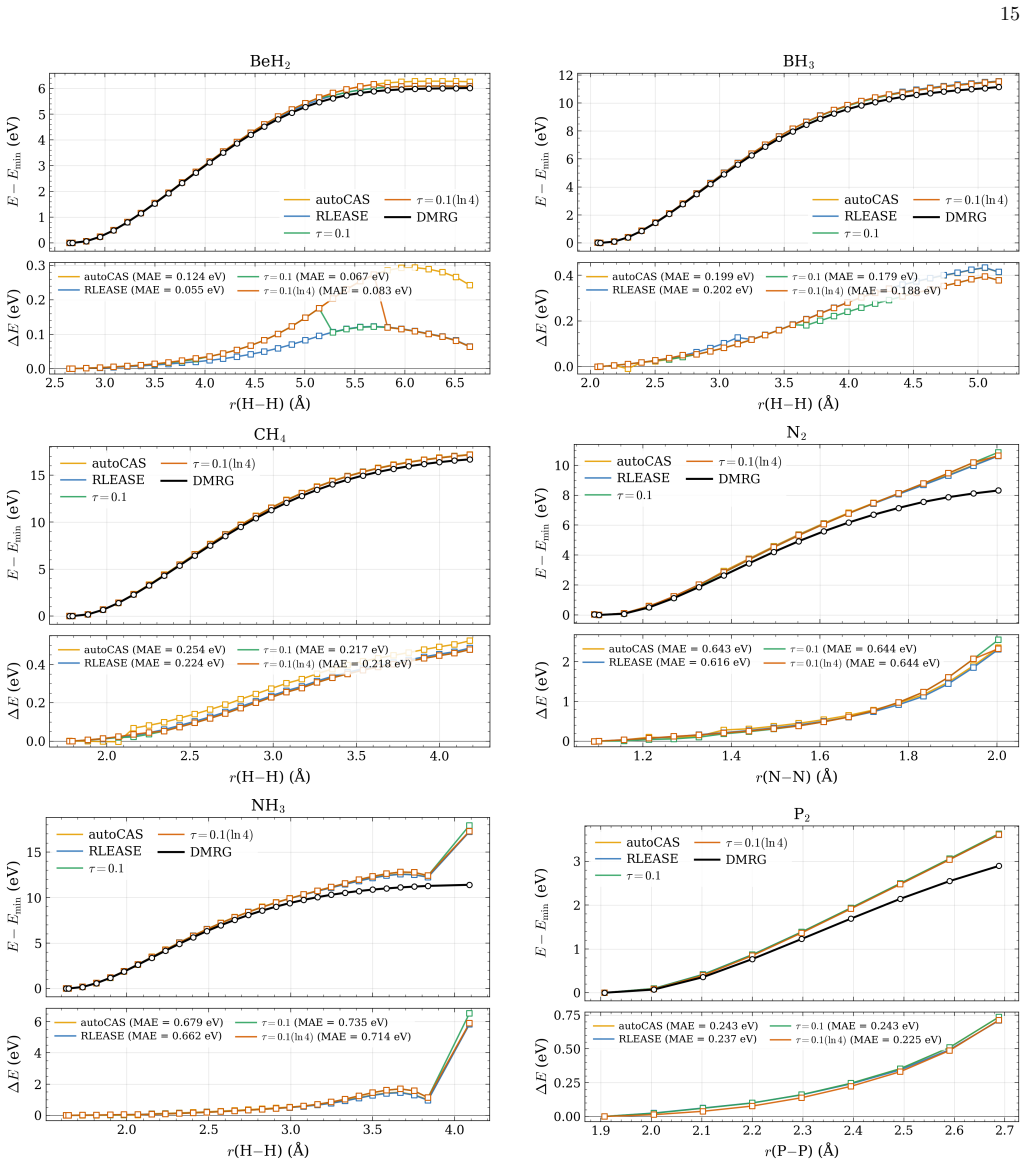
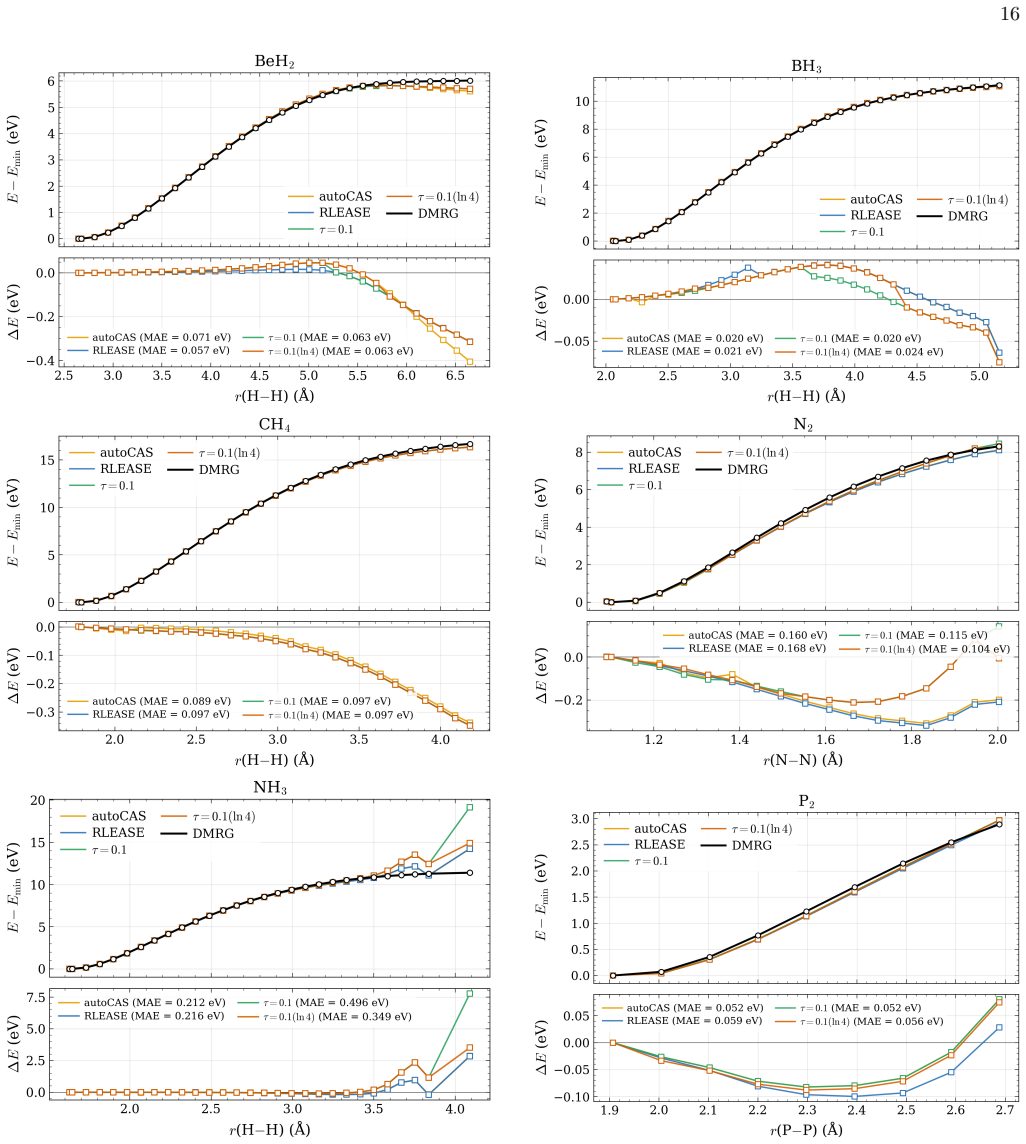
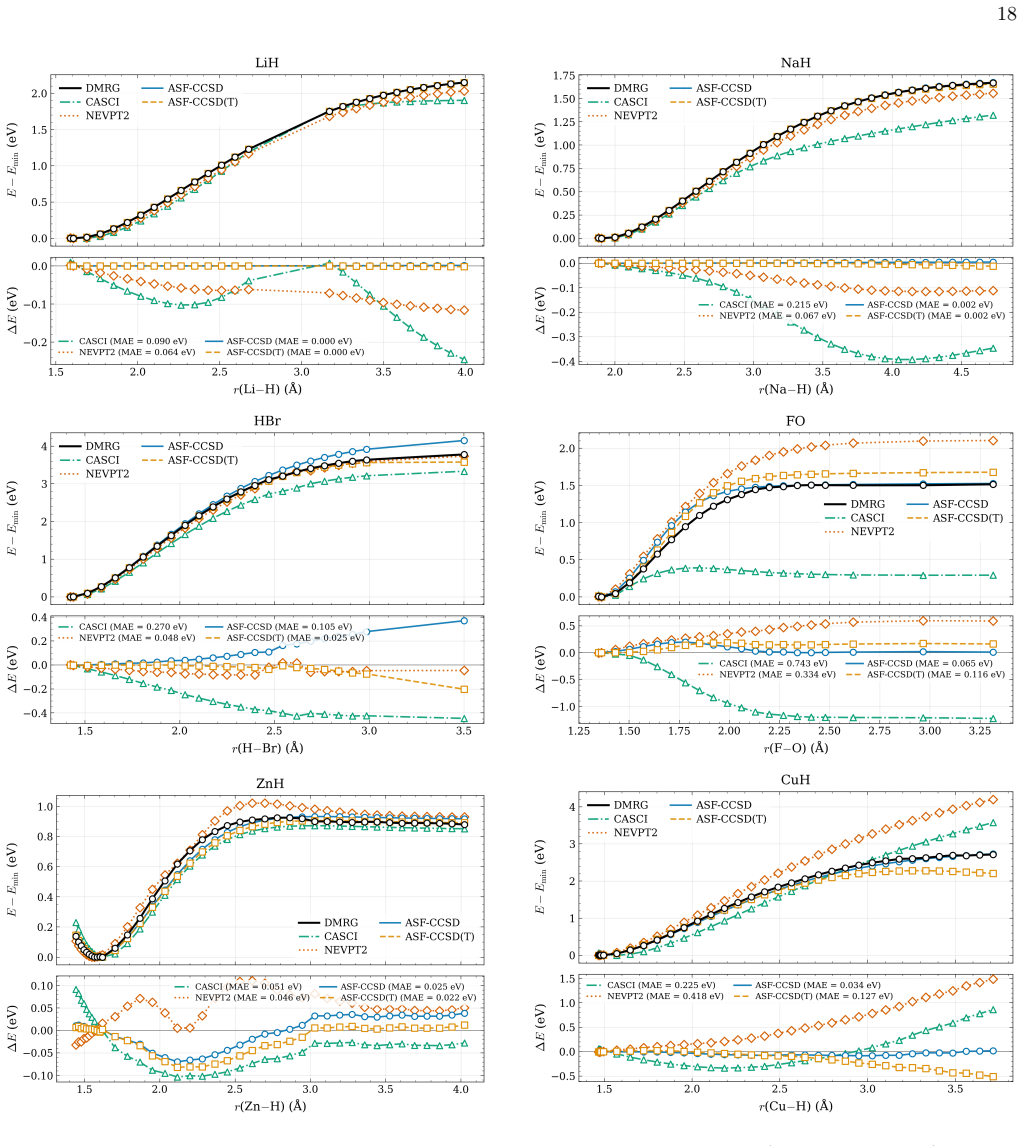
- [Abstract] Abstract: the claim that the learned threshold policy produces "competitive potential-energy surfaces relative to established entropy-based selectors" on test systems requires explicit quantitative support (e.g., error statistics or PES plots) demonstrating that the fixed network does not systematically select too many or too few orbitals when bonding types, spin states, or elements differ from the training set.
minor comments (1)
- Notation for the predicted per-orbital score (â§s_{1}) should be defined once and used consistently; the abstract introduces it without prior definition.
Simulated Author's Rebuttal
We thank the referee for the constructive and detailed comments. We address each major comment below and outline the revisions we will make to strengthen the manuscript.
read point-by-point responses
-
Referee: [Abstract] Abstract: the central transferability claim ("Despite being trained on a small set of molecules and geometries, RLEASE transfers to chemically diverse test systems") is load-bearing for the deployment narrative, yet the reward is defined directly as sc-NEVPT2–DMRG discrepancy; this creates an explicit dependence on the external DMRG benchmark during training that must be shown not to produce overfitting to the training distribution's electronic motifs.
Authors: We acknowledge that the reward function relies on DMRG reference energies during training, which could in principle lead to overfitting to motifs present in the training distribution. The manuscript already demonstrates transfer to chemically diverse test systems without retraining, but to more explicitly rule out overfitting we will add a dedicated subsection describing the chemical and electronic diversity of the training molecules and geometries, together with additional out-of-distribution performance metrics on the test set. revision: partial
-
Referee: [Abstract] Abstract: the claim that the learned threshold policy produces "competitive potential-energy surfaces relative to established entropy-based selectors" on test systems requires explicit quantitative support (e.g., error statistics or PES plots) demonstrating that the fixed network does not systematically select too many or too few orbitals when bonding types, spin states, or elements differ from the training set.
Authors: We agree that the competitiveness claim requires explicit quantitative backing. In the revised manuscript we will include tables reporting mean absolute errors and maximum deviations in sc-NEVPT2 energies relative to DMRG for both RLEASE and entropy-based selections across the full test set, as well as representative potential-energy-surface plots that illustrate active-space sizes and energy errors for molecules with bonding types, spin states, and elements outside the training distribution. revision: yes
Circularity Check
No significant circularity in the derivation or training pipeline.
full rationale
The PPO reward is the sc-NEVPT2 vs. DMRG discrepancy on a fixed training collection; this is an external oracle used to supervise the policy. Deployment applies the fixed network to new molecules using only HF descriptors, with no further DMRG or retraining. The transferability claim is an empirical assertion that can be (and is) tested on held-out systems rather than being true by construction. No self-citation is invoked to justify uniqueness or to close the loop, and no equation or definition reduces the output active-space selection to the training reward itself. The chain is therefore self-contained against external benchmarks.
Axiom & Free-Parameter Ledger
free parameters (1)
- neural-network weights and PPO policy parameters
axioms (2)
- domain assumption sc-NEVPT2 energy discrepancy with DMRG is a suitable scalar reward for active-space quality
- domain assumption Hartree-Fock orbital descriptors contain sufficient information to predict useful active-space membership
Reference graph
Works this paper leans on
-
[1]
B. O. Roos, R. Lindh, P. ˚A. Malmqvist, V. Veryazov, and P.-O. Widmark,Multiconfigurational Quantum Chemistry, 1st ed. (John Wiley & Sons, 2016)
2016
-
[2]
P. G. Szalay, T. M¨ uller, G. Gidofalvi, H. Lischka, and R. Shepard, Chemical Reviews112, 108 (2012), https://doi.org/10.1021/cr200137a
-
[3]
D. I. Lyakh, M. Musia l, V. F. Lotrich, and R. J. Bartlett, Chemical Reviews112, 182 (2012), https://doi.org/10.1021/cr2001417
-
[4]
B. O. Roos, P. R. Taylor, and P. E. Sigbahn, Chem. Phys.48, 157 (1980)
1980
-
[5]
P. E. M. Siegbahn, J. Alml¨ of, A. Heiberg, and B. O. Roos, J. Chem. Phys.74, 2384 (1981), https://doi.org/10.1063/1.441359
-
[6]
Siegbahn, A
P. Siegbahn, A. Heiberg, B. Roos, and B. Levy, Phys. Scr.21, 323 (1980)
1980
-
[7]
S. R. White, Physical Review Letters69, 2863 (1992)
1992
-
[8]
G. K.-L. Chan and S. Sharma, Annual Review of Physical Chemistry62, 465 (2011)
2011
-
[9]
R. J. Buenker and S. D. Peyerimhoff, Theoretica chimica acta35, 33 (1974)
1974
-
[10]
R. J. Buenker, S. D. Peyerimhoff, and W. Butscher, Mol. Phys.35, 771 (1978)
1978
-
[11]
Angeli, R
C. Angeli, R. Cimiraglia, S. Evangelisti, T. Leininger, and J.-P. Malrieu, Journal of Chemical Physics114, 10252 (2001)
2001
-
[12]
C. Angeli, S. Borini, M. Cestari, and R. Cimiraglia, J. Chem. Phys.121, 4043 (2004), https://doi.org/10.1063/1.1778711
-
[13]
Angeli, R
C. Angeli, R. Cimiraglia, and J.-P. Malrieu, Chem. Phys. Lett.350, 297 (2001)
2001
-
[14]
J. J. Wardzala, M. R. Hennefarth, V. Agarawal, B. Jangid, A. Seal, M. R. Hermes, D. S. King, and L. Gagliardi, Chemical Reviews126, 4592 (2026), pMID: 41930730, https://doi.org/10.1021/acs.chemrev.5c00866
-
[15]
Lloyd, Science273, 1073–1078 (1996)
S. Lloyd, Science273, 1073–1078 (1996)
1996
-
[16]
S. Verma, A. Mitra, Q. Wang, R. D’Cunha, B. Jangid, M. R. Hennefarth, V. Agarawal, L. Otis, S. Haldar, M. R. Hermes, and L. Gagliardi, Chemical Reviews126, 184 (2026), pMID: 41481354, https://doi.org/10.1021/acs.chemrev.5c00486
-
[17]
Veryazov, P
V. Veryazov, P. ˚A. Malmqvist, and B. O. Roos, International Journal of Quantum Chemistry111, 3329 (2011)
2011
-
[18]
Legeza and J
¨O. Legeza and J. S´ olyom, Physical Review B68, 195116 (2003)
2003
-
[19]
Rissler, R
J. Rissler, R. M. Noack, and S. R. White, Chemical Physics323, 519 (2006)
2006
-
[20]
C. J. Stein and M. Reiher, Journal of Chemical Theory and Computation12, 1760 (2016)
2016
-
[21]
E. R. Sayfutyarova, Q. Sun, G. K.-L. Chan, and G. Knizia, Journal of Chemical Theory and Computation13, 4063 (2017)
2017
-
[22]
Jeong, S
W. Jeong, S. J. Stoneburner, D. King, R. Li, A. Walker, R. Lindh, and L. Gagliardi, Journal of Chemical Theory and Computation16, 2389 (2020)
2020
-
[23]
Golub, A
P. Golub, A. Antalik, L. Veis, and J. Brabec, Journal of Chemical Theory and Computation17, 6053 (2021)
2021
-
[24]
Angeli, R
C. Angeli, R. Cimiraglia, and J.-P. Malrieu, Journal of Chemical Physics117, 9138 (2002)
2002
-
[25]
Sunet al., WIREs Computational Molecular Science8, e1340 (2018)
Q. Sunet al., WIREs Computational Molecular Science8, e1340 (2018)
2018
-
[26]
R. J. Bartlett and M. Musia l, Reviews of Modern Physics79, 291 (2007)
2007
-
[27]
Raghavachari, G
K. Raghavachari, G. W. Trucks, J. A. Pople, and M. Head-Gordon, Chemical Physics Letters157, 479 (1989)
1989
-
[28]
D. S. King and L. Gagliardi, Journal of Chemical Theory and Computation17, 2817 (2021)
2021
-
[29]
D. S. King, M. R. Hermes, D. G. Truhlar, and L. Gagliardi, Journal of Chemical Theory and Computation18, 6065 (2022)
2022
-
[30]
J. Schulman, F. Wolski, P. Dhariwal, A. Radford, and O. Klimov, arXiv preprint arXiv:1707.06347 (2017)
Pith/arXiv arXiv 2017
-
[31]
Zhai and G
H. Zhai and G. K.-L. Chan, Journal of Chemical Physics154, 224116 (2021)
2021
-
[32]
H. Zhai, H. R. Larsson, S. Lee, Z.-H. Cui, T. Zhu, C. Sun,et al., Journal of Chemical Physics159, 234801 (2023)
2023
-
[33]
A. E. Clark and E. R. Davidson, J. Org. Chem.68, 3387 (2003)
2003
-
[34]
Batatia, D
I. Batatia, D. P. Kov´ acs, G. N. C. Simm, C. Ortner, and G. Cs´ anyi, inAdvances in Neural Information Processing Systems, Vol. 35 (2022)
2022
-
[35]
Batzner, A
S. Batzner, A. Musaelian, L. Sun, M. Geiger, J. P. Mailoa, M. Kornbluth, N. Molinari, T. E. Smidt, and B. Kozinsky, Nature Communications13, 2453 (2022)
2022
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.