Querying Counterfactuals on Tissue Graphs with Supervised Disentanglement
Pith reviewed 2026-06-27 17:43 UTC · model grok-4.3
The pith
Supervised disentanglement separates a cell's intrinsic state from its spatial neighbors to answer counterfactual queries on tissue graphs.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
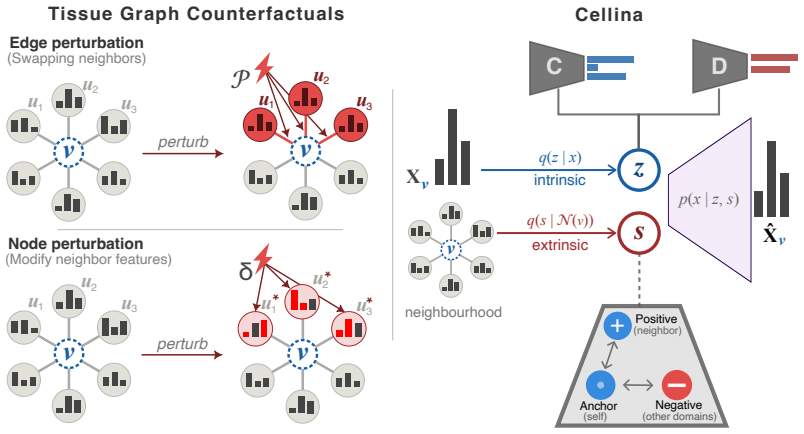
Tissue graph counterfactuals are formalized as two families of spatial interventions: edge perturbations that rewire neighbor relations and node perturbations that alter neighbor expression. Supervised disentanglement decomposes each cell into an intrinsic state vector and a spatial context vector; the spatial vector is then treated as a controllable conditioning variable so that new neighbor configurations produce predicted expression changes for the target cell.
What carries the argument
Supervised disentanglement that isolates a cell's intrinsic state from its spatial context so the spatial component can serve as a conditioning input for counterfactual prediction.
If this is right
- The method outperforms both spatially aware and non-spatial baselines on in-silico edge and node perturbations across millions of cells.
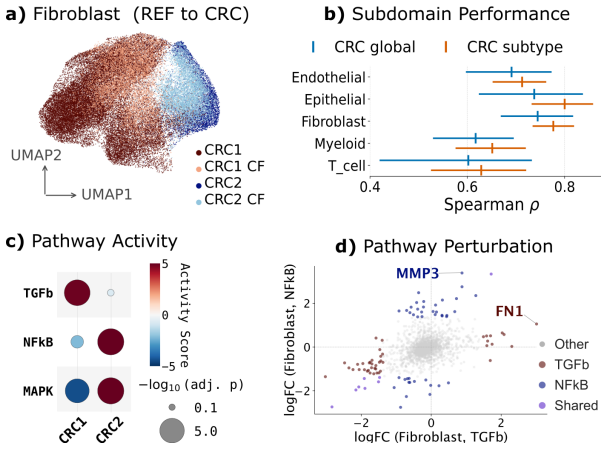
- Unsupervised clustering on the learned intrinsic states recovers biologically distinct cancer subdomains.
- The framework supports direct simulation of targeted changes to a cell's neighbor set.
- Runtime and memory scale to tissue graphs containing millions of spatially resolved cells.
Where Pith is reading between the lines
- The same intrinsic-versus-context split could be tested on time-series spatial data to track how neighbor changes drive state transitions during disease progression.
- If the separation holds, the model could be paired with perturbation screens to prioritize which neighbor alterations are most likely to shift a target cell's phenotype.
- Extending the conditioning variable to include additional modalities such as chromatin state or metabolite levels would test whether spatial context alone captures the dominant drivers of expression change.
Load-bearing premise
Supervised disentanglement cleanly isolates a cell's intrinsic state from its spatial context without leaking biologically relevant information or degrading prediction accuracy.
What would settle it
Collect a new spatial dataset containing cells whose actual post-perturbation expression is known; run the model on the pre-perturbation graphs and check whether its predicted counterfactual expressions match the observed post-perturbation values within measurement error.
Figures

read the original abstract
Tissue graph counterfactuals ask how a cell's expression would change under altered spatial neighbor contexts. Such queries are central to predicting cell behavior in tissues, but lack a unified definition, with existing methods targeting specific intervention types or treating cells as i.i.d. In this work, we first formalize tissue graph counterfactuals as a class of spatial interventions that either rewire connections between cells (edge perturbation) or modify the expression of their neighbors (node perturbation). We then introduce Cellina (https://cellina.readthedocs.io) - a framework that uses supervised disentanglement to decompose a cell's intrinsic state from its spatial context, using the latter as a conditioning input for counterfactual predictions. Across benchmarks spanning over 2.5 million spatially-resolved cells in colorectal cancer and mouse brain, Cellina outperforms spatially-informed and non-spatial competitors in in-silico graph perturbations, disentanglement, and scalability. Additionally, we show that Cellina reveals biologically distinct cancer subdomains in an unsupervised manner and enables targeted neighbor perturbation simulations.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript formalizes tissue graph counterfactuals as a class of spatial interventions consisting of edge perturbations (rewiring cell connections) or node perturbations (modifying neighbor expression). It introduces Cellina, a framework that applies supervised disentanglement to decompose each cell's intrinsic state from its spatial context and uses the spatial component as a conditioning input for counterfactual predictions. On benchmarks covering >2.5 million spatially resolved cells from colorectal cancer and mouse brain, Cellina is reported to outperform both spatially-informed and non-spatial baselines in in-silico perturbation accuracy, disentanglement quality, and scalability; additional results show unsupervised identification of biologically distinct cancer subdomains and targeted neighbor-perturbation simulations.
Significance. If the central claims hold, the work supplies a unified, scalable approach to counterfactual reasoning on tissue graphs, addressing a gap between existing methods that target narrow intervention types or ignore spatial structure. The scale of the evaluated datasets and the demonstration of unsupervised subdomain discovery constitute concrete strengths. The supervised disentanglement strategy, if shown to avoid leakage, could become a reusable technique for spatial transcriptomics.
major comments (2)
- [Abstract] Abstract: the central claim that supervised disentanglement cleanly separates intrinsic state from spatial context (enabling leakage-free conditioning for counterfactuals) is load-bearing for all reported performance gains, yet the abstract supplies no quantitative validation, ablation, or leakage diagnostic; without these, the outperformance cannot be assessed.
- [Abstract] Abstract: the statement that Cellina 'outperforms' competitors across in-silico graph perturbations, disentanglement, and scalability is presented without reference to error bars, data-split protocols, number of replicates, or statistical tests; this information is required to evaluate whether the reported superiority is robust.
minor comments (1)
- The provision of a documentation link (cellina.readthedocs.io) is helpful for reproducibility and should be retained.
Simulated Author's Rebuttal
We thank the referee for the constructive comments regarding the abstract. The abstract is intended as a concise summary of the work, with full quantitative details, protocols, and diagnostics provided in the main text and supplementary materials. We address each point below and indicate where revisions can be made.
read point-by-point responses
-
Referee: [Abstract] Abstract: the central claim that supervised disentanglement cleanly separates intrinsic state from spatial context (enabling leakage-free conditioning for counterfactuals) is load-bearing for all reported performance gains, yet the abstract supplies no quantitative validation, ablation, or leakage diagnostic; without these, the outperformance cannot be assessed.
Authors: The abstract summarizes the core contribution without space for full metrics. The manuscript provides quantitative validation of the disentanglement (via performance on counterfactual tasks, ablation studies removing the spatial component, and leakage checks through cross-validation of intrinsic vs. context factors) in the Results and Methods sections, supported by experiments on >2.5M cells. We can revise the abstract to include a brief clause referencing these validations (e.g., 'validated via ablations and leakage diagnostics on large-scale benchmarks') if the editor permits an expanded abstract. revision: partial
-
Referee: [Abstract] Abstract: the statement that Cellina 'outperforms' competitors across in-silico graph perturbations, disentanglement, and scalability is presented without reference to error bars, data-split protocols, number of replicates, or statistical tests; this information is required to evaluate whether the reported superiority is robust.
Authors: The abstract reports the high-level outcome; the full experimental details—including multiple data splits, replicate counts, error bars (standard deviations across runs), and statistical tests (e.g., paired t-tests or Wilcoxon tests)—are documented in the Methods, Results, and Supplementary sections. We can add a short parenthetical in the abstract (e.g., 'with statistical significance across replicates') or a reference to the supplementary materials to address this. revision: partial
Circularity Check
No significant circularity detected
full rationale
The provided abstract and description formalize tissue graph counterfactuals as edge/node perturbations and introduce Cellina via supervised disentanglement for conditioning predictions. No equations, derivations, or self-citations are exhibited that reduce any claimed prediction or result to fitted inputs or prior author work by construction. The central claims rest on external benchmark evaluations across 2.5M cells rather than internal self-definition or renaming. The derivation chain is self-contained against the stated benchmarks with no load-bearing circular steps identifiable from the text.
Axiom & Free-Parameter Ledger
Reference graph
Works this paper leans on
-
[1]
How attentive are graph attention networks?arXiv preprint arXiv:2105.14491,
Shaked Brody, Uri Alon, and Eran Yahav. How attentive are graph attention networks?arXiv preprint arXiv:2105.14491,
-
[2]
doi: 10.1016/j.cell.2024.11.015
ISSN 0092-8674. doi: 10.1016/j.cell.2024.11.015. URLhttps://doi.org/10.1016/j.cell.2024.11.015. Helena L Crowell, Irene Ruano, Zedong Hu, Yourae Hong, Gin Caratù, Hubert Piessevaux, Ash- ley Heck, Rachel Liu, Max Walter, Megan Vandenberg, et al. Tracing colorectal malignancy transformation from cell to tissue scale.bioRxiv,
-
[3]
Fast graph representation learning with pytorch geometric.arXiv preprint arXiv:1903.02428,
11 Matthias Fey and Jan Eric Lenssen. Fast graph representation learning with pytorch geometric.arXiv preprint arXiv:1903.02428,
Pith/arXiv arXiv 1903
-
[4]
Strand: Sequence-conditioned transport for single-cell perturbations
Boyang Fu, George Dasoulas, Sameer Gabbita, Xiang Lin, Shanghua Gao, Xiaorui Su, Soumya Ghosh, and Marinka Zitnik. Strand: Sequence-conditioned transport for single-cell perturbations. arXiv preprint arXiv:2602.10156,
-
[5]
Demystifying inter-class disentanglement.arXiv preprint arXiv:1906.11796,
Aviv Gabbay and Yedid Hoshen. Demystifying inter-class disentanglement.arXiv preprint arXiv:1906.11796,
arXiv 1906
-
[6]
Ilyes Khemakhem, Diederik Kingma, Ricardo Monti, and Aapo Hyvarinen
URL https://proceedings.neurips.cc/paper_files/paper/2022/ file/aa933b5abc1be30baece1d230ec575a7-Paper-Conference.pdf. Ilyes Khemakhem, Diederik Kingma, Ricardo Monti, and Aapo Hyvarinen. Variational autoencoders and nonlinear ica: A unifying framework. InInternational conference on artificial intelligence and statistics, pages 2207–2217. PMLR,
2022
-
[8]
Diederik P Kingma and Max Welling
URLhttps://arxiv.org/abs/2004.11362. Diederik P Kingma and Max Welling. Auto-encoding variational bayes.arXiv preprint arXiv:1312.6114,
arXiv 2004
-
[9]
Variational graph auto-encoders.arXiv preprint arXiv:1611.07308,
Thomas N Kipf and Max Welling. Variational graph auto-encoders.arXiv preprint arXiv:1611.07308,
-
[10]
doi: 10.1038/s41592-021-01336-8
ISSN 1548-7105. doi: 10.1038/s41592-021-01336-8. URLhttps://doi.org/10.1038/s41592-021-01336-8. Jing Ma, Ruocheng Guo, Saumitra Mishra, Aidong Zhang, and Jundong Li. Clear: Generative counterfactual explanations on graphs.Advances in neural information processing systems, 35: 25895–25907,
-
[11]
doi: 10.52202/079017-0650. URL https://proceedings.neurips.cc/paper_files/paper/2024/file/ 24c4d51f3ef48dd2dbab78243ecb26a1-Paper-Datasets_and_Benchmarks_Track.pdf. Jovan Tanevski, Ricardo Omar Ramirez Flores, Attila Gabor, Denis Schapiro, and Julio Saez- Rodriguez. Explainable multiview framework for dissecting spatial relationships from highly multiplex...
-
[12]
Yan Wu, Esther Wershof, Sebastian M Schmon, Marcel Nassar, Bła˙zej Osi´nski, Ridvan Eksi, Zichao Yan, Rory Stark, Kun Zhang, and Thore Graepel. Perturbench: Benchmarking machine learning models for cellular perturbation analysis.arXiv preprint arXiv:2408.10609,
-
[13]
Let c(obs) g,v denote the observed raw count of gene g in cell v, and c(pred) g,v the corresponding model-predicted raw count. We convert both to normalized expression as pg,v = cg,vP g′ cg′,v ·ℓ 0 We consider perturbation settings where each gene is evaluated between a control and a perturbed condition. The log-fold change (logFC) for genegis defined as ...
arXiv 2024
-
[14]
Bio Conservation
were not used in our analyses for the following reasons: slide 110 contained sequencing artefacts in the form of major empty patches disrupting neighborhood computations, while slide 222 did not contain any REF cells. For evaluations, we merged fine-grained subtypes (e.g., epithelial subpopulations annotated as Epi1-Epi4) into broad cell type categories. ...
2023
-
[15]
with subsampling to 103, 104 and 105 cells (Figure A6). For a fair comparison, we set the same batch size and number of epochs for each model and only compare the train loop (without pre-processing workloads).Cellinacomes out as one of the fastest-to-train models in the benchmark suite, owing to the efficient pseudobulk-based φ(v) computed a priori, omitt...
2023
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.