When Three-Dimensional Conformer Ensembles Improve Molecular Property Prediction Beyond Two-Dimensional Fingerprints: A Systematic Study
Pith reviewed 2026-06-27 17:33 UTC · model grok-4.3
The pith
Conformer ensemble statistics improve predictions for solvation properties but not electronic or steric ones.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
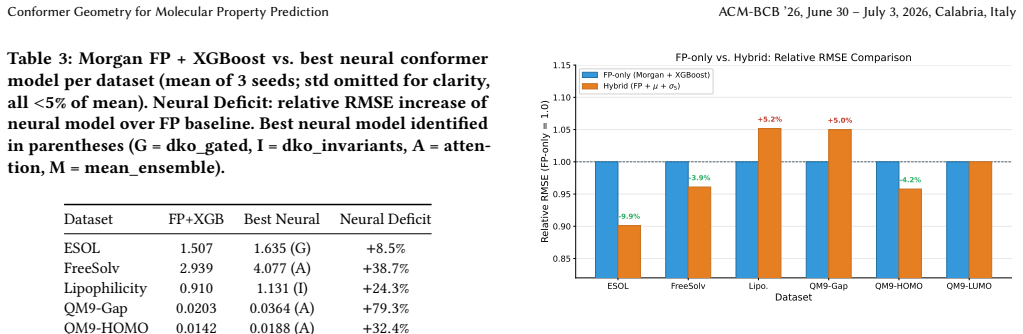
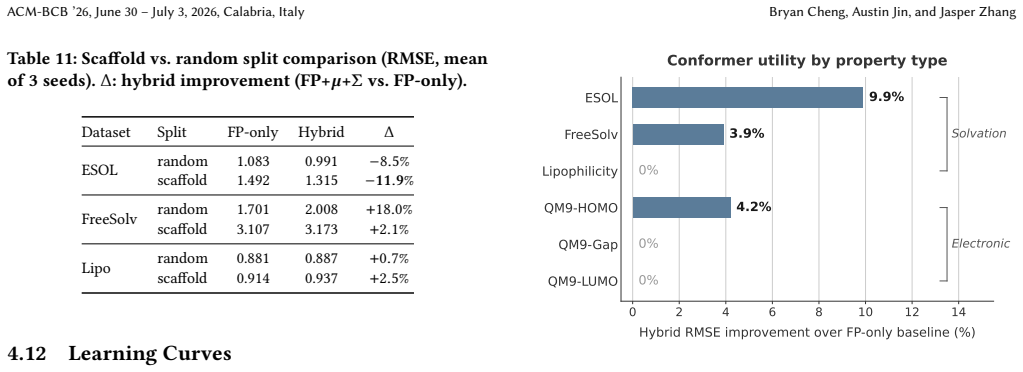
Conformer ensemble statistics extracted via Distribution Kernel Operators yield statistically significant RMSE reductions on solvation-dependent properties (ESOL -11.0 percent, p less than 10 to the minus 9; FreeSolv -13.5 percent, p less than 3 times 10 to the minus 5) while providing no benefit for electronic or steric tasks. Three lines of evidence confirm the selectivity has a physical basis: larger gains under scaffold splits than random splits, concentration on large flexible molecules, and monotonic growth with training set size. A four-tier performance hierarchy places end-to-end 3D graph neural networks highest, followed by engineered physicochemical descriptors, then Morgan fingerp
What carries the argument
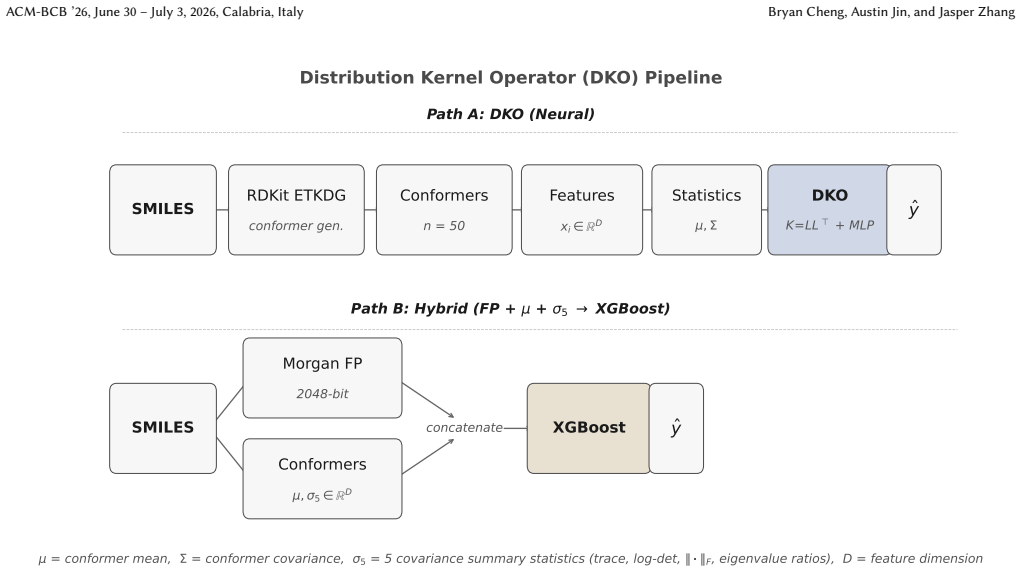
Distribution Kernel Operators that extract mean and covariance statistics from conformer ensembles and feed them as features.
If this is right
- Solvation-dependent properties gain from conformer statistics while electronic and steric properties do not.
- Simple scalar invariants from conformer means outperform all tested covariance architectures.
- Pre-computed feature approaches are limited by an information bottleneck compared with end-to-end 3D graph neural networks.
- Improvement concentrates on larger, more flexible molecules under scaffold splits.
Where Pith is reading between the lines
- For applications focused on solubility or hydration, adding conformer generation may be worth the cost, while for many other molecular tasks it is not.
- The finding that covariance features add almost no signal suggests future ensemble methods could drop covariance computation entirely.
- The hierarchy implies that when computational budget allows, training a 3D graph neural network end-to-end is preferable to any pipeline that freezes conformer features first.
Load-bearing premise
The assumption that the observed difference in performance across property types reflects real physical mechanisms rather than dataset biases or modeling artifacts.
What would settle it
Repeating the same experiments on an independent solvation dataset that shows no error reduction, or on an electronic property dataset that shows a reduction, would falsify the selectivity claim.
Figures

read the original abstract
When do three-dimensional conformer ensembles improve molecular property prediction beyond two-dimensional fingerprints? We provide the first systematic, mechanistically grounded answer. Through ~1,000 experiments spanning 13 model configurations, 14 regression targets, and 2 classification targets across MoleculeNet, QM9, and MARCEL benchmarks, we discover selective complementarity: conformer ensemble statistics extracted via Distribution Kernel Operators (DKOs) yield statistically significant RMSE reductions on solvation-dependent properties (ESOL -11.0%, p < 10^{-9}; FreeSolv -13.5%, p < 3x10^{-5}; 10-seed paired validation) while providing no benefit for electronic or steric tasks. Three lines of evidence confirm this selectivity has a physical rather than statistical basis: improvement is larger under scaffold splits than random splits (+11.9% vs. +8.5% on ESOL), concentrates on large, flexible molecules (+18.9% for heaviest quartile), and grows monotonically with training data. We establish a four-tier performance hierarchy: end-to-end 3D GNNs (SchNet, PaiNN; 21-42% over fingerprints) >= engineered physicochemical descriptors (PMI/SASA/USR) > Morgan fingerprints + XGBoost > all neural conformer ensemble methods, confirmed by two architecturally diverse GNNs and revealing that the pre-computed feature bottleneck limits ensemble approaches. Feature attribution and mutual information analysis expose the mechanistic asymmetry: conformer mean features carry 2-8x more information per feature than fingerprint bits, yet covariance features contribute <2% of model signal, explaining why five simple scalar invariants outperform all complex covariance architectures (p < 0.001). These findings yield an empirical property taxonomy and a practical decision framework for when conformer generation is worth the investment.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript reports results from approximately 1000 experiments across 13 model configurations, 16 targets from MoleculeNet/QM9/MARCEL, and multiple splits, claiming that Distribution Kernel Operators applied to 3D conformer ensembles yield statistically significant RMSE reductions specifically on solvation-dependent properties (ESOL -11.0% p<10^{-9}; FreeSolv -13.5% p<3x10^{-5}) but none on electronic or steric tasks. It further claims this selectivity has a physical basis based on larger gains under scaffold splits, concentration in the heaviest MW quartile (+18.9%), and monotonic improvement with training set size, while establishing a performance hierarchy with end-to-end 3D GNNs at the top and neural conformer methods at the bottom, and identifying that mean features dominate over covariance features.
Significance. If the empirical improvements and hierarchy hold after verification of methods, the work supplies a practical decision framework and property taxonomy for when conformer generation is worthwhile in molecular ML pipelines. The scale of the benchmarking, the use of paired validation, and the feature attribution analysis that quantifies the 2-8x information advantage of mean features are positive contributions. The finding that five scalar invariants outperform complex covariance architectures is a useful negative result.
major comments (1)
- [Abstract] Abstract: The assertion that the observed selectivity 'has a physical rather than statistical basis' is supported by three comparisons (scaffold vs. random split gains, MW-quartile concentration, monotonic data-size growth). However, these comparisons do not rule out confounding by flexibility (e.g., rotatable-bond count or conformer diversity), which may correlate with both scaffold membership and solvation targets; the abstract gives no indication that the analyses were stratified or matched on such metrics.
minor comments (1)
- The abstract states results from '10-seed paired validation' and 'two architecturally diverse GNNs' but does not name the GNNs or detail exclusion rules/hyperparameter search; the main text should make these explicit to allow reproduction of the reported p-values.
Simulated Author's Rebuttal
We thank the referee for their constructive feedback. We address the single major comment below.
read point-by-point responses
-
Referee: [Abstract] Abstract: The assertion that the observed selectivity 'has a physical rather than statistical basis' is supported by three comparisons (scaffold vs. random split gains, MW-quartile concentration, monotonic data-size growth). However, these comparisons do not rule out confounding by flexibility (e.g., rotatable-bond count or conformer diversity), which may correlate with both scaffold membership and solvation targets; the abstract gives no indication that the analyses were stratified or matched on such metrics.
Authors: We thank the referee for identifying this gap in our supporting analyses. The three comparisons were selected because they align with physical expectations for solvation (larger, more flexible molecules under scaffold splits benefit more), but we acknowledge that explicit stratification or matching on rotatable-bond count or conformer diversity was not performed and could represent a confounder. We will revise the abstract to qualify the claim, stating that the evidence is consistent with a physical basis while noting the lack of flexibility controls as a limitation. We will also add a supplementary stratification of gains by rotatable-bond count (and, where data permit, by conformer diversity) to test robustness. This is a partial revision that clarifies interpretation without altering the reported empirical results or hierarchy. revision: partial
Circularity Check
Empirical benchmarking study; no derivation reduces to inputs
full rationale
This is a large-scale empirical study reporting RMSE reductions, p-values, and split/quartile comparisons from ~1000 experiments on MoleculeNet/QM9/MARCEL. All load-bearing claims (selective complementarity for solvation properties, performance hierarchy, mechanistic asymmetry via feature attribution) are direct outputs of the benchmark runs and statistical tests. No equations, fitted parameters, or self-citations are invoked to derive the reported improvements; the results stand as independent measurements against external benchmarks.
Axiom & Free-Parameter Ledger
Reference graph
Works this paper leans on
-
[1]
Pedro J Ballester and W Graham Richards. 2007. Ultrafast Shape Recognition to Search Compound Databases for Similar Molecular Shapes.Journal of Computa- tional Chemistry28, 10 (2007), 1711–1723
2007
-
[2]
Guy W Bemis and Mark A Murcko. 1996. The Properties of Known Drugs. 1. Molecular Frameworks.Journal of Medicinal Chemistry39, 15 (1996), 2887–2893
1996
-
[3]
BioRender. 2025. BioRender.com. https://biorender.com Scientific figure created with BioRender.com
2025
-
[4]
Tianqi Chen and Carlos Guestrin. 2016. XGBoost: A Scalable Tree Boosting System. InProceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining. 785–794
2016
-
[5]
John S Delaney. 2004. ESOL: Estimating Aqueous Solubility Directly from Molec- ular Structure.Journal of Chemical Information and Computer Sciences44, 3 (2004), 1000–1005
2004
-
[6]
Octavian-Eugen Ganea, Lagnajit Pattanaik, Connor W Coley, Regina Barzilay, Klavs F Jensen, William H Green, and Tommi S Jaakkola. 2021. GeoMol: Torsional Geometric Generation of Molecular 3D Conformer Ensembles. InAdvances in Neural Information Processing Systems, Vol. 34
2021
-
[7]
Johannes Gasteiger, Shankari Giri, Johannes T Margraf, and Stephan Günne- mann. 2020. Fast and Uncertainty-Aware Directional Message Passing for Non- Equilibrium Molecules. InMachine Learning for Molecules Workshop at NeurIPS
2020
-
[8]
Johannes Gasteiger, Janek Groß, and Stephan Günnemann. 2020. Directional Message Passing for Molecular Graphs. InInternational Conference on Learning Representations
2020
-
[9]
Tobias Gensch, Gabriel dos Passos Gomes, Pascal Friederich, Ellyn Peters, Theresa Gauber, Joseph R Struble, Spencer D Dreher, Franziska Schoenebeck, Matthew S Sigman, and Alán Aspuru-Guzik. 2022. A Comprehensive Discovery Platform for Organophosphorus Ligands for Catalysis.Journal of the American Chemical Society144, 3 (2022), 1205–1217
2022
-
[10]
Justin Gilmer, Samuel S Schoenholz, Patrick F Riley, Oriol Vinyals, and George E Dahl. 2017. Neural Message Passing for Quantum Chemistry. InInternational Conference on Machine Learning. 1263–1272
2017
-
[11]
Paul C D Hawkins. 2017. Conformation Generation: The State of the Art.Journal of Chemical Information and Modeling57, 8 (2017), 1747–1756
2017
-
[12]
Paul C D Hawkins, A Geoffrey Skillman, Gregory L Warren, Benjamin A Ellingson, and Matthew T Stahl. 2010. Conformer Generation with OMEGA: Algorithm and Validation Using High Quality Structures from the Protein Databank and Cambridge Structural Database.Journal of Chemical Information and Modeling 50, 4 (2010), 572–584
2010
-
[13]
Esther Heid, Kevin P Greenman, Yujie Chung, Shih-Cheng Li, David E Graff, Florence H Vermeire, Haoyang Wu, William H Green, and Charles J McGill
-
[14]
Journal of Chemical Information and Modeling64, 1 (2024), 9–17
Chemprop: A Machine Learning Package for Chemical Property Prediction. Journal of Chemical Information and Modeling64, 1 (2024), 9–17
2024
-
[15]
Jarmo Huuskonen. 2000. Estimation of Aqueous Solubility for a Diverse Set of Organic Compounds Based on Molecular Topology.Journal of Chemical Information and Computer Sciences40, 3 (2000), 773–777. ACM-BCB ’26, June 30 – July 3, 2026, Calabria, Italy Bryan Cheng, Austin Jin, and Jasper Zhang
2000
-
[16]
Dejun Jiang, Zhenxing Wu, Chang-Yu Hsieh, Guangyong Chen, Ben Liao, Zhe Wang, Chao Shen, Dongsheng Cao, Jian Wu, and Tingjun Hou. 2021. Could Graph Neural Networks Learn Better Molecular Representation for Drug Discovery? A Comparison Study of Descriptor-based and Graph-based Models.Journal of Cheminformatics13, 1 (2021), 12
2021
-
[17]
Diederik P Kingma and Jimmy Ba. 2015. Adam: A Method for Stochastic Opti- mization. InInternational Conference on Learning Representations
2015
-
[18]
Alexander Kraskov, Harald Stögbauer, and Peter Grassberger. 2004. Estimating Mutual Information.Physical Review E69, 6 (2004), 066138
2004
-
[19]
Gregory A Landrum. 2016. RDKit: Open-Source Cheminformatics Software. (2016). https://www.rdkit.org
2016
-
[20]
Olivier Ledoit and Michael Wolf. 2004. A well-conditioned estimator for large- dimensional covariance matrices.Journal of Multivariate Analysis88, 2 (2004), 365–411
2004
-
[21]
Juho Lee, Yoonho Lee, Jungtaek Kim, Adam R Kosiorek, Seungjin Choi, and Yee Whye Teh. 2019. Set Transformer: A Framework for Attention-based Permutation-Invariant Input. InInternational Conference on Machine Learning
2019
-
[22]
Yi-Lun Liao and Tess Smidt. 2023. Equiformer: Equivariant Graph Attention Transformer for 3D Atomistic Graphs. InInternational Conference on Learning Representations
2023
-
[23]
Christopher A Lipinski, Franco Lombardo, Beryl W Dominy, Paul J Feeney, et al
-
[24]
Experimental and Computational Approaches to Estimate Solubility and Permeability in Drug Discovery and Development Settings.Advanced Drug Delivery Reviews46, 1–3 (2001), 3–26
2001
-
[25]
Ilya Loshchilov and Frank Hutter. 2019. Decoupled Weight Decay Regularization. International Conference on Learning Representations(2019)
2019
-
[26]
Simon Mitternacht. 2016. FreeSASA: An Open Source C Library for Solvent Accessible Surface Area Calculations.F1000Research5 (2016), 189
2016
-
[27]
David L Mobley and J Peter Guthrie. 2014. FreeSolv: A Database of Experimental and Calculated Hydration Free Energies, with Input Files.Journal of Computer- Aided Molecular Design28, 7 (2014), 711–720
2014
-
[28]
Philipp Pracht, Fabian Bohle, and Stefan Grimme. 2020. Automated Exploration of the Low-Energy Chemical Space with Fast Quantum Chemical Methods.Physical Chemistry Chemical Physics22, 14 (2020), 7169–7192
2020
-
[29]
Raghunathan Ramakrishnan, Pavlo O Dral, Matthias Rupp, and O Anatole von Lilienfeld. 2014. Quantum Chemistry Structures and Properties of 134 Kilo Molecules.Scientific Data1 (2014), 140022
2014
-
[30]
Sereina Riniker and Gregory A Landrum. 2015. Better Informed Distance Geom- etry: Using What We Know To Improve Conformation Generation.Journal of Chemical Information and Modeling55, 12 (2015), 2562–2574
2015
-
[31]
David Rogers and Mathew Hahn. 2010. Extended-Connectivity Fingerprints. Journal of Chemical Information and Modeling50, 5 (2010), 742–754
2010
-
[32]
Yu Rong, Yatao Bian, Tingyang Xu, Weiyang Xie, Ying Wei, Wenbing Huang, and Junzhou Huang. 2020. Self-Supervised Graph Transformer on Large-Scale Molecular Data. InAdvances in Neural Information Processing Systems, Vol. 33
2020
-
[33]
Matthias Rupp, Alexandre Tkatchenko, Klaus-Robert Müller, and O Anatole von Lilienfeld. 2012. Fast and Accurate Modeling of Molecular Atomization Energies with Machine Learning.Physical Review Letters108, 5 (2012), 058301
2012
-
[34]
Kristof T Schütt, Pieter-Jan Kindermans, Huziel Enoc Sauceda, Stefan Chmiela, Alexandre Tkatchenko, and Klaus-Robert Müller. 2017. SchNet: A Continuous- Filter Convolutional Neural Network for Modeling Quantum Interactions. In Advances in Neural Information Processing Systems, Vol. 30
2017
-
[35]
Kristof T Schütt, Oliver T Unke, and Michael Gastegger. 2021. Equivariant Message Passing for the Prediction of Tensorial Properties and Molecular Spectra. InInternational Conference on Machine Learning. 9377–9388
2021
-
[36]
Hannes Stark, Dominique Beaini, Gabriele Corso, Prudencio Tossou, Christian Dallago, Stephan Günnemann, and Pietro Liò. 2022. 3D Infomax Improves GNNs for Molecular Property Prediction. InInternational Conference on Machine Learn- ing
2022
-
[37]
B L Welch. 1947. The Generalization of ‘Student’s’ Problem when Several Differ- ent Population Variances are Involved.Biometrika34, 1/2 (1947), 28–35
1947
-
[38]
Zhenqin Wu, Bharath Ramsundar, Evan N Feinberg, Joseph Gomes, Caleb Ge- niesse, Aneesh S Pappu, Karl Leswing, and Vijay Pande. 2018. MoleculeNet: A Benchmark for Molecular Machine Learning.Chemical Science9, 2 (2018), 513–530
2018
-
[39]
Kevin Yang, Kyle Swanson, Wengong Jin, Connor Coley, Philipp Eiden, Hua Gao, Angel Guzman-Perez, Timothy Hopper, Brian Kelley, Miriam Mathea, et al. 2019. Analyzing Learned Molecular Representations for Property Prediction.Journal of Chemical Information and Modeling59, 8 (2019), 3370–3388
2019
-
[40]
Manzil Zaheer, Satwik Kottur, Siamak Ravanbakhsh, Barnabas Poczos, Ruslan R Salakhutdinov, and Alexander J Smola. 2017. Deep Sets. InAdvances in Neural Information Processing Systems, Vol. 30
2017
-
[41]
Gengmo Zhou, Zhifeng Gao, Qiankun Ding, Hang Zheng, Hongteng Xu, Zhewei Wei, Linfeng Zhang, and Guolin Ke. 2023. Uni-Mol: A Universal 3D Molecu- lar Representation Learning Framework.International Conference on Learning Representations(2023)
2023
-
[42]
3D” = 28 physicochemical descriptors; “Geo
Yanqiao Zhu, Jeehyun Hwang, Keir Adams, Zhen Liu, Bozhao Nan, Brock Stenfors, Yuanqi Du, Jatin Chauhan, Olaf Wiest, Olexandr Isayev, Connor W Coley, Yizhou Sun, and Wei Wang. 2024. Learning Over Molecular Conformer Ensembles: Datasets and Benchmarks. InInternational Conference on Learning Representa- tions. Conformer Geometry for Molecular Property Predic...
2024
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.