Molecular dynamic simulation of multicomponent CoCrFeNiMn high-entropy alloy thin film deposition
Pith reviewed 2026-06-27 16:02 UTC · model grok-4.3
The pith
Molecular dynamics simulation of CoCrFeMnNi high-entropy alloy film deposition reproduces experimental phase composition and lattice parameters.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
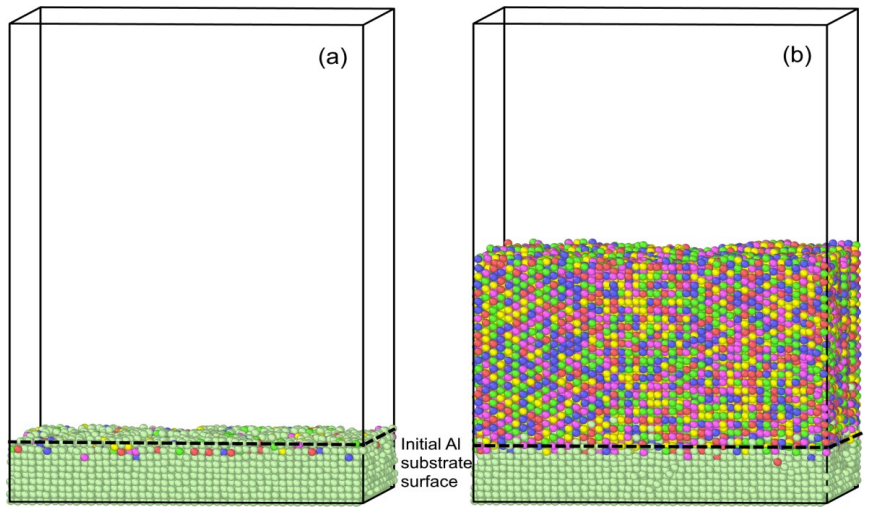
During a 100 ns molecular dynamics simulation, 50,000 atoms of the CoCrFeMnNi high-entropy alloy were deposited onto an Al(100) substrate using Morse potentials with mixing rules, forming a film approximately 6.1 nm thick that contains FCC, BCC, HCP, and amorphous regions; the phase composition and structural parameters determined from radial distribution functions agree with experimental data.
What carries the argument
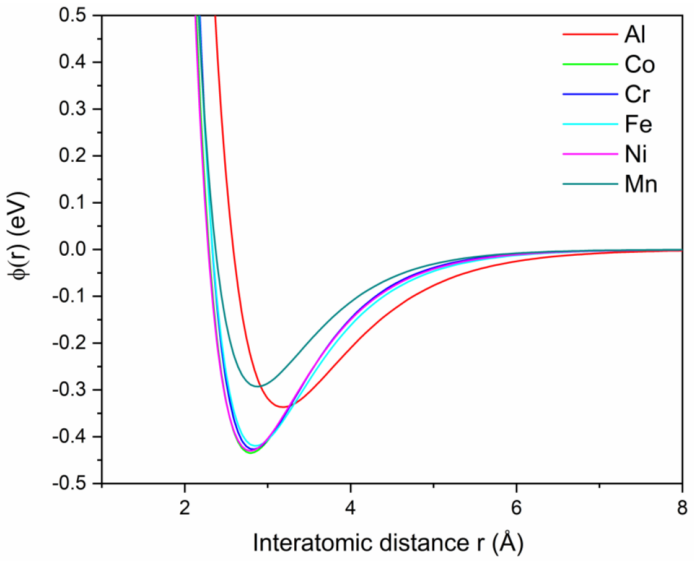
Calibrated Morse potentials combined with mixing rules for regular solutions that model interatomic interactions in the multicomponent alloy system.
If this is right
- The method allows prediction of film structure for similar high-entropy alloys without physical experiments.
- Radial distribution function analysis provides estimates of lattice parameters for crystalline phases in the simulated film.
- The coexistence of multiple crystal structures and amorphous regions is a feature of the deposited film.
- Validation against experiment supports use of this potential set for deposition simulations.
Where Pith is reading between the lines
- Such simulations could be extended to test how changing deposition energy affects the phase balance in the film.
- Results may inform design of high-entropy alloy coatings for wear or corrosion resistance by predicting microstructure.
- Comparison with other potential types could show if Morse potentials are sufficient or if more advanced models are needed for accuracy.
Load-bearing premise
The calibrated Morse potentials with mixing rules for regular solutions accurately describe the interatomic interactions during the deposition process.
What would settle it
An experiment that deposits a CoCrFeMnNi film under similar conditions and measures significantly different phase fractions or lattice constants would falsify the simulation's predictive accuracy.
Figures
read the original abstract
The deposition and growth of a thin CoCrFeMnNi high-entropy alloy film on an Al(100) substrate were investigated by molecular dynamics simulation. Interatomic interactions were described using a calibrated set of Morse potentials combined with mixing rules for regular solutions. During a 100 ns simulation, 50,000 atoms with an incident energy of 10 eV were deposited, producing a film of about 6.1 nm thickness. The resulting film contains face-centred cubic (FCC), body-centred cubic (BCC), hexagonal close-packed (HCP), and amorphous regions. Analysis of the radial distribution function (RDF) was used to determine nearest-neighbour distances and estimate lattice parameters for the crystalline phases. The simulated phase composition and structural parameters are in good agreement with available experimental data.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript reports molecular dynamics simulations of CoCrFeMnNi high-entropy alloy thin film deposition on an Al(100) substrate. Interatomic interactions are modeled with a calibrated set of Morse potentials combined with regular-solution mixing rules. A 100 ns run deposits 50,000 atoms at 10 eV incident energy, yielding a ~6.1 nm film containing FCC, BCC, HCP, and amorphous regions. Nearest-neighbor distances and lattice parameters are extracted from the radial distribution function, and the authors state that the simulated phase composition and structural parameters agree with available experimental data.
Significance. If the interatomic potentials are shown to be reliable, the simulation provides atomistic detail on the kinetics of phase selection during energetic deposition of a five-component HEA, a regime where in-situ experimental probes are limited. The work could serve as a starting point for exploring deposition-parameter effects on microstructure, provided the model is first validated against independent properties such as cohesive energies, elastic constants, or known binary/ternary phase stabilities.
major comments (3)
- [Abstract] Abstract: the central claim that 'the simulated phase composition and structural parameters are in good agreement with available experimental data' is presented without any quantitative metrics (e.g., percentage deviations in lattice parameters, phase-fraction comparisons, or root-mean-square errors). This absence prevents assessment of whether the agreement is meaningful or merely qualitative.
- [Methods / Interatomic potentials] The description of the interatomic potentials (Morse form plus mixing rules): no calibration details, target properties, or validation against known Co-Cr-Fe-Mn-Ni formation energies, stacking-fault energies, or FCC/BCC/HCP relative stabilities are supplied. Because the headline result (phase mixture) is produced by these potentials, the lack of such benchmarks makes the agreement with experiment difficult to interpret as a prediction rather than a consequence of the chosen functional form.
- [Results] Results section on phase identification: the assignment of FCC, BCC, HCP, and amorphous regions appears to rest solely on visual inspection and RDF peak positions. No order-parameter analysis (e.g., common-neighbor analysis, bond-orientational order) or quantitative comparison of simulated versus experimental lattice constants with uncertainties is reported, weakening the structural-parameter agreement claim.
minor comments (2)
- [Methods] The manuscript should specify the exact number of independent runs, thermostat/barostat settings, and substrate temperature to allow reproducibility.
- [Results] Figure captions and text should clarify how the ~6.1 nm thickness was measured and whether it includes the substrate-film interface.
Simulated Author's Rebuttal
We thank the referee for the constructive comments on our manuscript. We address each major point below and will revise the manuscript to improve clarity and rigor where appropriate.
read point-by-point responses
-
Referee: [Abstract] Abstract: the central claim that 'the simulated phase composition and structural parameters are in good agreement with available experimental data' is presented without any quantitative metrics (e.g., percentage deviations in lattice parameters, phase-fraction comparisons, or root-mean-square errors). This absence prevents assessment of whether the agreement is meaningful or merely qualitative.
Authors: We agree that quantitative metrics are needed to substantiate the agreement claim. In the revised manuscript we will add explicit comparisons, including percentage deviations for lattice parameters extracted from the RDF and phase-fraction estimates relative to the cited experimental data. revision: yes
-
Referee: [Methods / Interatomic potentials] The description of the interatomic potentials (Morse form plus mixing rules): no calibration details, target properties, or validation against known Co-Cr-Fe-Mn-Ni formation energies, stacking-fault energies, or FCC/BCC/HCP relative stabilities are supplied. Because the headline result (phase mixture) is produced by these potentials, the lack of such benchmarks makes the agreement with experiment difficult to interpret as a prediction rather than a consequence of the chosen functional form.
Authors: The Morse potentials were calibrated to experimental cohesive energies and lattice constants of the pure metals and selected binaries, but we acknowledge that the manuscript omitted the specific calibration targets and any further validation. We will expand the Methods section to document the calibration procedure and include available comparisons to formation energies or relative phase stabilities from the literature. revision: yes
-
Referee: [Results] Results section on phase identification: the assignment of FCC, BCC, HCP, and amorphous regions appears to rest solely on visual inspection and RDF peak positions. No order-parameter analysis (e.g., common-neighbor analysis, bond-orientational order) or quantitative comparison of simulated versus experimental lattice constants with uncertainties is reported, weakening the structural-parameter agreement claim.
Authors: Phase assignment was based on RDF peak positions together with direct visualization of atomic environments. We agree that quantitative order-parameter analysis would strengthen the results. In the revision we will apply common-neighbor analysis to obtain phase fractions and will report uncertainties on the lattice parameters derived from the RDF. revision: yes
Circularity Check
No circularity: MD simulation validated against external experimental data using standard calibrated potentials
full rationale
The paper describes a standard molecular dynamics deposition simulation on an Al substrate using a pre-calibrated set of Morse pair potentials plus regular-solution mixing rules. The output (phase fractions, RDF-derived lattice parameters after 100 ns deposition) is compared directly to independent experimental measurements on CoCrFeMnNi films. No step reduces the reported agreement to a fitted parameter by construction, no self-citation supplies a load-bearing uniqueness theorem, and the calibration of the potentials is treated as an input rather than derived from the deposition results themselves. The derivation chain is therefore self-contained against external benchmarks.
Axiom & Free-Parameter Ledger
free parameters (1)
- Morse potential parameters
axioms (1)
- domain assumption Morse potentials with mixing rules for regular solutions accurately model interatomic forces in CoCrFeMnNi during deposition
Reference graph
Works this paper leans on
-
[1]
D.B. Miracle and O.N. Senkov, Acta Mater., 122, 448 (2017). doi:10.1016/j.actamat.2016.08.081
-
[2]
Y. Zhou, H. Xiang and F.-Z. Dai, High‐Entropy Materials. From Basics to Applications. 1st ed. Weinheim, Germany: Wiley, (2023). doi:10.1002/9783527837205
-
[3]
J. Brechtl and P.K. Liaw, High-Entropy Materials: Theory, Experiments, and Applications. Edited by J. Brechtl and P.K. Liaw. Cham: Springer International Publishing, (2021). doi:10.1007/978-3-030-77641-1
-
[4]
O.I. Kushnerov and V.F. Bashev East Eur. J. Phys., 25 (3), 43 (2021). doi:10.26565/2312-4334-2021-3-06
-
[5]
V.A. Polonskyy, V.F. Bashev and O.I. Kushnerov J. Chem. Technol., 28 (2), 176 (2020). doi:10.15421/082019
-
[6]
V.A. Polonskyy, V.F. Bashev and O.I. Kushnerov J. Chem. Technol., 30 (1), 88 (2022). doi:10.15421/jchemtech.v30i1.237109
-
[7]
G.S. Firstov et al. Prog. Phys. Met., 24 (4), 819 (2023). doi:10.15407/ufm.24.04.819
-
[8]
V. Girzhon, V. Yemelianchenko and O. Smolyakov Acta Metall. Slovaca, 29 (1), 44 (2023). doi:10.36547/ams.29.1.1710
-
[9]
Two-dimensional Sentiment Analysis of text
O.I. Kushnerov, S.I. Ryabtsev and V.F. Bashev Mol. Cryst. Liq. Cryst., 750 (1), 135 (2023). doi:10.1080/15421406.2022.2073043
work page internal anchor Pith review Pith/arXiv arXiv doi:10.1080/15421406.2022.2073043 2023
-
[10]
V.F. Bashev, O.I. Kushnerov and S.I. Ryabtsev Mol. Cryst. Liq. Cryst., 765 (1), 145 (2023). doi:10.1080/15421406.2023.2215125
work page internal anchor Pith review Pith/arXiv arXiv doi:10.1080/15421406.2023.2215125 2023
-
[11]
I. Shtablavyi et al. Phys. Chem. Solid State, 25 (1), 5 (2024). doi:10.15330/pcss.25.1.5-13
-
[12]
W.-M. Choi et al. Npj Comput. Mater., 4 (1), 1 (2018). doi:10.1038/s41524-017- 0060-9
-
[13]
R. Gröger, V. Vitek and A. Dlouhý Model. Simul. Mater. Sci. Eng., 28 (7), 075006 (2020). doi:10.1088/1361-651X/ab7f8b
-
[14]
C. Varvenne et al. Phys. Rev. B, 93 (10), 104201 (2016). doi:10.1103/PhysRevB.93.104201
-
[15]
A.P. Thompson et al. Comput. Phys. Commun., 271, 108171 (2022). doi:10.1016/j.cpc.2021.108171
-
[16]
W.M. Brown et al. Comput. Phys. Commun., 182 (4), 898 (2011). doi:10.1016/J.CPC.2010.12.021
-
[17]
Deviations from Tribimaximal Neutrino Mixing using a Model with $\Delta(27)$ Symmetry
W.M. Brown et al. Comput. Phys. Commun., 183 (3), 449 (2012). doi:10.1016/J.CPC.2011.10.012. This is an Accepted Manuscript of an article published in Molecular Crystals and Liquid Crystals, Volume 769, Issues 7–8, 2025, pp. 762–772. The Version of Record is available at: https://doi.org/10.1080/15421406.2025.2504044
work page internal anchor Pith review Pith/arXiv arXiv doi:10.1016/j.cpc.2011.10.012 2012
-
[18]
A. Stukowski Model. Simul. Mater. Sci. Eng., 18 (1), 015012 (2010). doi:10.1088/0965-0393/18/1/015012
-
[19]
V.-T. Nguyen, V. Thi Thu Nhu and X.-T. Vo J. Cryst. Growth, 603, 127004 (2023). doi:10.1016/j.jcrysgro.2022.127004
-
[20]
A. Liang et al. Acta Mater., 257, 119163 (2023). doi:10.1016/j.actamat.2023.119163
-
[21]
L. Xie et al. Appl. Surf. Sci., 285 (PART B), 810 (2013). doi:10.1016/j.apsusc.2013.08.133
-
[22]
O.I. Kushnerov, V.F. Bashev and S.I. Ryabtsev Springer Proc. Phys., 263, 419 (2021). doi:10.1007/978-3-030-74741-1_28
-
[23]
D.W. Jacobson and G.B. Thompson Comput. Mater. Sci., 205 (January), (2022). doi:10.1016/j.commatsci.2022.111206
-
[24]
B. Lv et al. Metals (Basel)., 12 (6), 982 (2022). doi:10.3390/met12060982
-
[25]
P.T.M. Hanh et al. Results Phys., 19, 103632 (2020). doi:10.1016/j.rinp.2020.103632
-
[26]
M. Lindroos et al. Chem. Phys. Lett., 173 (1), 92 (1990). doi:10.1016/0009- 2614(90)85309-Z
-
[27]
I. Aslam et al. Materialia, 8 (September), 100473 (2019). doi:10.1016/j.mtla.2019.100473
-
[28]
G. Bonny et al. J. Nucl. Mater., 442 (1–3), 282 (2013). doi:10.1016/j.jnucmat.2013.08.018
-
[29]
https://www.webelements.com
-
[30]
S.K. Das, D. Roy and S. Sengupta J. Phys. F Met. Phys., 7 (1), 5 (1977). doi:10.1088/0305-4608/7/1/011
-
[31]
X. Sun et al. Acta Mater., 140, (2017). doi:10.1016/j.actamat.2017.08.045
-
[32]
J.Y.Y. He et al. Acta Mater., 62 (1), 105 (2014). doi:10.1016/j.actamat.2013.09.037
-
[33]
H. Zhang et al. Acta Mater., 155, 12 (2018). doi:10.1016/j.actamat.2018.05.050
-
[34]
J. Kumar et al. Acta Mater., 238, 118208 (2022). doi:10.1016/j.actamat.2022.118208
-
[35]
Y. Qi, X. Chen and M. Feng Mater. Sci. Eng. A, 791 (May), 139444 (2020). doi:10.1016/j.msea.2020.139444
-
[36]
F. Zhang et al. Nat. Commun., 8 (1), 15687 (2017). doi:10.1038/ncomms15687
-
[37]
R. Chulist et al. Materials (Basel), 15 (23), 8407 (2022). doi:10.3390/ma15238407
-
[38]
F. Zhang et al. Entropy, 21 (3), 239 (2019). doi:10.3390/e21030239
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.