Flexible Kernels for Protein Property Prediction
Pith reviewed 2026-06-27 14:13 UTC · model grok-4.3
The pith
Sequence kernels from evolutionary matrices and local linearity yield data-efficient Gaussian processes for protein properties.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
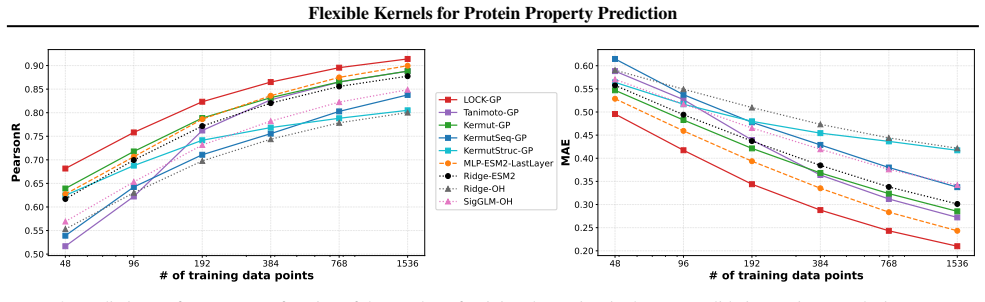
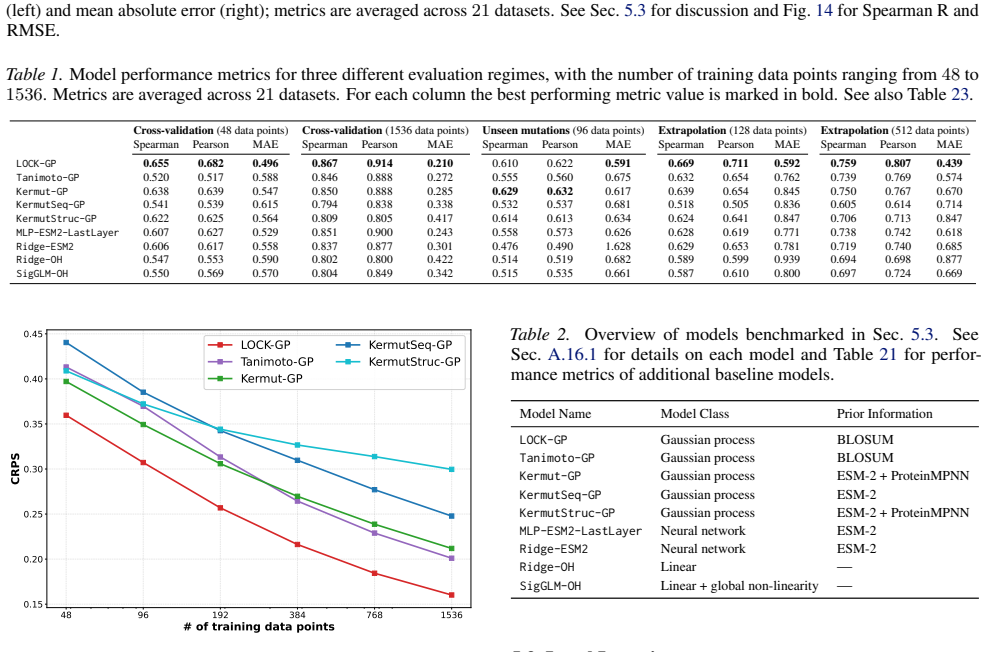
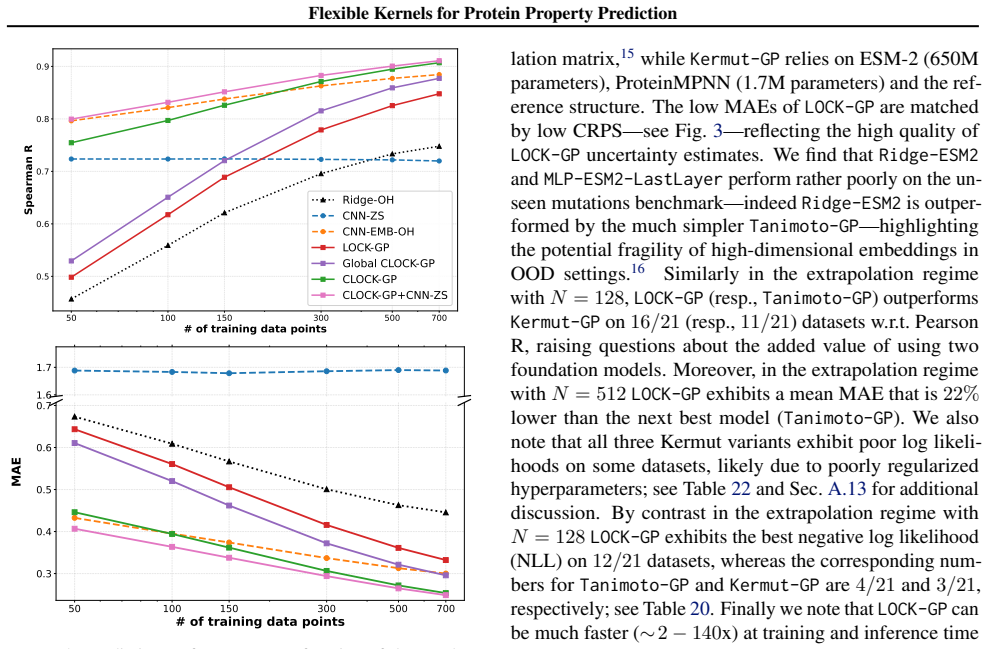
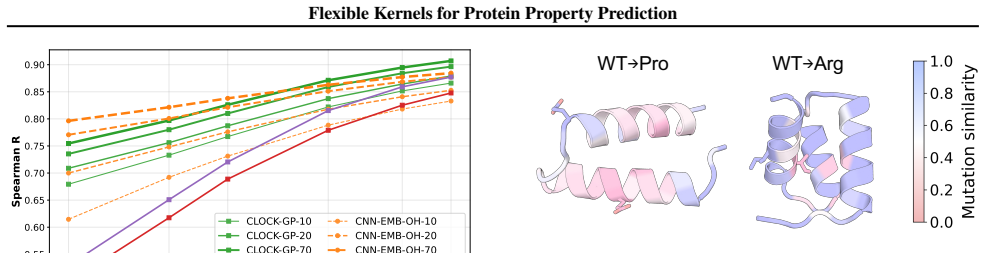
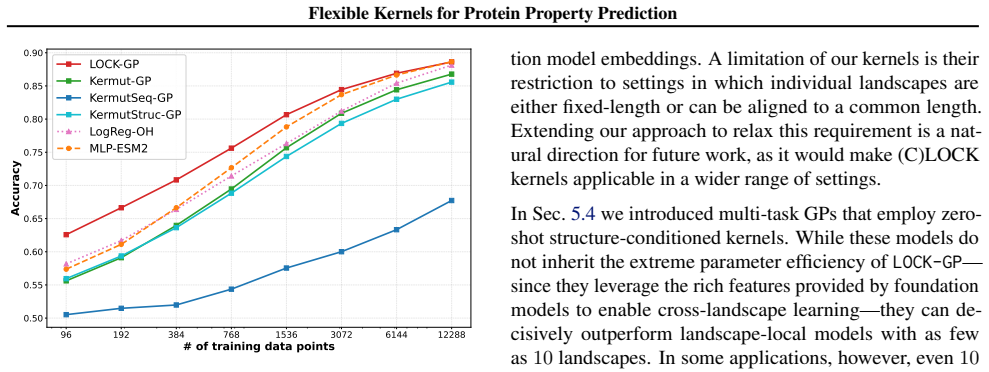
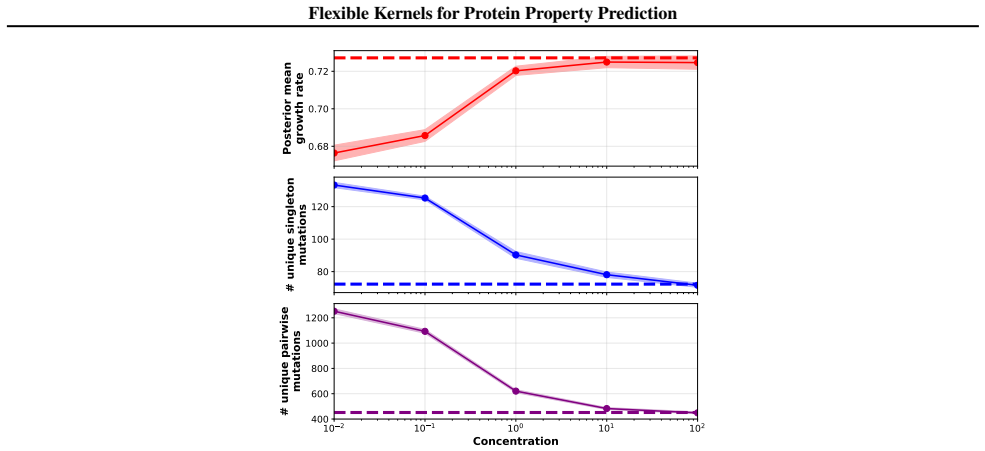
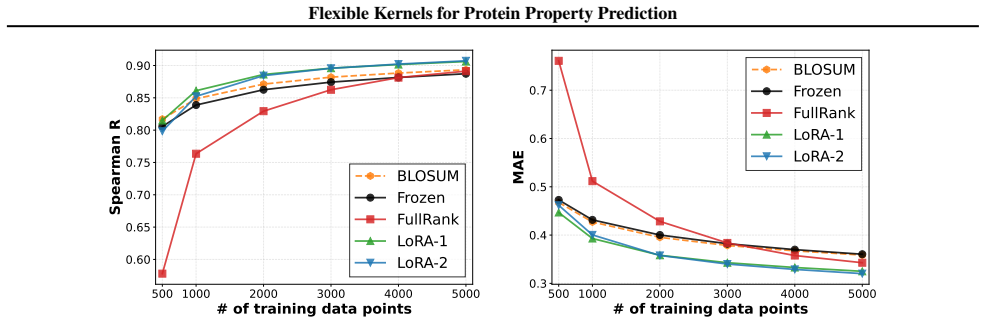
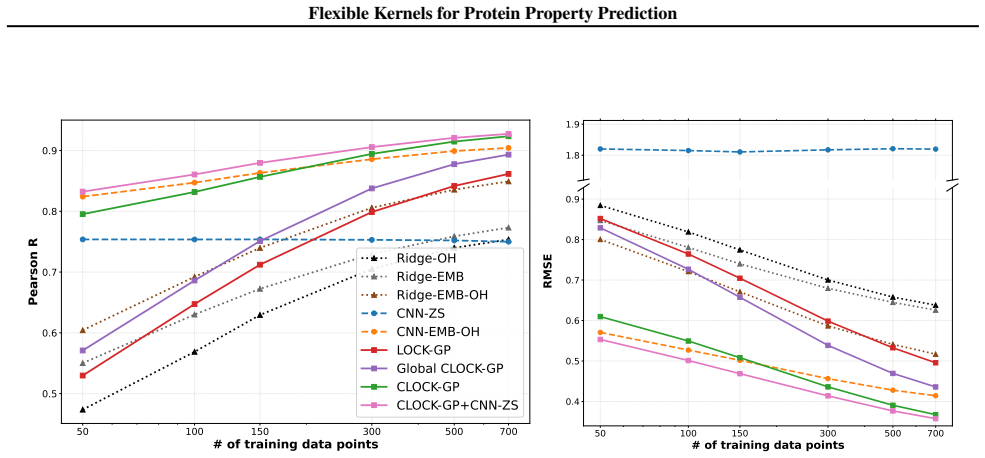
We introduce a class of sequence kernels that exploit evolutionary substitution matrices as well as local linearity and demonstrate that the resulting Gaussian processes provide data-efficient models of protein property landscapes, frequently outperforming alternatives that rely on foundation model embeddings. Furthermore--by learning what are in effect structure-aware substitution matrices--we show that our kernels can readily incorporate structural information from foundation models. We demonstrate that these structure-conditioned kernels are well suited to multi-task learning across multiple protein property landscapes and can decisively outperform local supervised learning methods.
What carries the argument
Sequence kernels that combine evolutionary substitution matrices with local linearity, which can be conditioned on structural data to learn structure-aware substitution matrices.
Load-bearing premise
Kernels built from evolutionary substitution matrices plus local linearity generalize across protein families and properties without requiring extensive hyperparameter tuning or suffering from distribution shift when applied to new sequences.
What would settle it
A comparison on held-out protein families or properties where the Gaussian process models built from these kernels fail to match or exceed the predictive accuracy or data efficiency of foundation-model embedding baselines.
Figures



read the original abstract
Despite its importance to applications in protein design, predicting protein properties like binding affinity and thermostability from sparse experimental data remains a significant challenge. Accordingly, we introduce a class of sequence kernels that exploit evolutionary substitution matrices as well as local linearity and demonstrate that the resulting Gaussian processes provide data-efficient models of protein property landscapes, frequently outperforming alternatives that rely on foundation model embeddings. Furthermore--by learning what are in effect structure-aware substitution matrices--we show that our kernels can readily incorporate structural information from foundation models. We demonstrate that these structure-conditioned kernels are well suited to multi-task learning across multiple protein property landscapes and can decisively outperform local supervised learning methods.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript introduces a class of sequence kernels for Gaussian processes that exploit evolutionary substitution matrices together with local linearity to model protein property landscapes from sparse data. It further shows how these kernels can be conditioned on structural information from foundation models by learning structure-aware substitution matrices. The central empirical claims are that the resulting GPs are data-efficient and frequently outperform alternatives based on foundation model embeddings, and that the structure-conditioned kernels are particularly well-suited to multi-task learning across multiple protein properties, where they can decisively outperform local supervised learning methods.
Significance. If the reported performance gains are robust, the work would offer a useful kernel-based alternative to embedding-heavy approaches for low-data protein property prediction tasks such as binding affinity and thermostability. The explicit incorporation of evolutionary priors and the multi-task capability are strengths that could reduce reliance on large foundation models while remaining computationally tractable. The approach appears to operate with few or no free parameters beyond the kernel construction itself.
minor comments (1)
- [Abstract] Abstract: the strong claims of frequent outperformance and decisive multi-task gains would be easier to evaluate if the abstract briefly indicated the protein families, number of properties/tasks, and evaluation protocol used.
Simulated Author's Rebuttal
We thank the referee for the positive assessment of our manuscript and the recommendation for minor revision. The summary accurately reflects the core contributions regarding sequence kernels that leverage substitution matrices and local linearity for data-efficient Gaussian process modeling of protein properties, as well as the structure-aware extensions for multi-task learning.
Circularity Check
No significant circularity identified
full rationale
The provided abstract and context describe an empirical introduction of sequence kernels exploiting substitution matrices and local linearity, with Gaussian process performance claims presented as experimental outcomes on protein property data. No derivation chain, equations, or self-citations are visible that reduce predictions to fitted inputs by construction, self-definitional loops, or load-bearing self-citations. The central claims rest on outperformance demonstrations rather than tautological redefinitions, making the work self-contained against external benchmarks with no circular steps exhibited.
Axiom & Free-Parameter Ledger
Reference graph
Works this paper leans on
-
[1]
URL http: //dx.doi.org/10.1038/s41467-024-45621-4
doi: 10.1038/s41467-024-45621-4. URL http: //dx.doi.org/10.1038/s41467-024-45621-4. Elnaggar, A., Heinzinger, M., Dallago, C., Rehawi, G., Wang, Y ., Jones, L., Gibbs, T., Feher, T., Angerer, C., Steinegger, M., Bhowmik, D., and Rost, B. Prottrans: Toward understanding the language of life through self- supervised learning.IEEE Transactions on Pattern Ana...
-
[2]
Highly accurate protein structure prediction with
doi: 10.1038/s41586-021-03819-2. Khan, A., Cowen-Rivers, A. I., Grosnit, A., Deik, D.-G.- X., Robert, P. A., Greiff, V ., Smorodina, E., Rawat, P., Akbar, R., Dreczkowski, K., et al. Toward real-world automated antibody design with combinatorial bayesian optimization.Cell Reports Methods, 3(1), 2023. Koshi, J. M. and Goldstein, R. A. Context-dependent opt...
-
[3]
ISSN 2050-084X. doi: 10.7554/elife.83442. URL http://dx.doi.org/10.7554/eLife.83442. Notin, P., Kollasch, A., Ritter, D., Van Niekerk, L., Paul, S., Spinner, H., Rollins, N., Shaw, A., Orenbuch, R., Weitz- man, R., et al. Proteingym: Large-scale benchmarks for protein fitness prediction and design.Advances in Neu- ral Information Processing Systems, 36:64...
-
[4]
for everyt >0, the kernelk t(i, j) = exp −t ψ(i, j) is positive semidefinite
-
[5]
for finite index sets,ψis a squared Euclidean distance. A.1.2. CHARACTERIZINGINFINITELYDIVISIBLEMATRICES With these definitions in hand, we can give a precise characterization of infinitely divisible matrices. Lemma A.8(Berg et al. (1984)).Let K be a symmetric matrix with strictly positive entries. Then K is infinitely divisible if and only iflog ◦ Kiscon...
1984
-
[6]
linear kernel plus non-linear kernel times linear kernel
There exist pointsx 1, . . . , xA inR m (for somem≤A−1) such that Kij = exp − ∥xi −x j∥2 for alli, j. In particular, every infinitely divisible correlation matrix is of the formK= exp ◦(−D) where D is asquared Euclidean distance matrix. Proof. By Lemma A.8,(1) ⇔ (2). By Schoenberg’s Theorem A.7,(2) ⇔ (3): D is CNSD iff e−tD is PSD for all t >0 , and for f...
1977
-
[7]
prior on each ˜αℓ.Marginally, this choice corresponds to a LogNormal(0, 5
-
[8]
prior on αℓ. We note that choosing an overcomplete parameterization has an impact on theoptimization dynamics; in particular by introducing ˜αwe expect it to be easier to take larger steps in αℓ space. These priors were chosen based on regression experiments conducted with datasets disjoint from those used in our experiments in Sec. 5. A.4. Epistasis and ...
2016
-
[9]
non-unique) sequences
it heavily penalizes sets of sequencesx 1:N that contain duplicate (i.e. non-unique) sequences
-
[10]
catastrophic NLL
it strongly encourages the sequences x1:N to spread out in a balanced manner across a nested set of 8 Hamming shells centered around the wild-type sequencex wt In particular Ψ(x1:N) strongly encourages exactly 100 of the 800 sequences to reside in the2-Hamming shell of xwt, exactly 100 of the 800 sequences to reside in the 3-Hamming shell of xwt, and so o...
2010
-
[11]
PLM UsageESM-2 and ESM-1v were pre-trained on single chain sequences, while some of our datasets consist of multi-chain proteins
is used instead of ESM-2-8M. PLM UsageESM-2 and ESM-1v were pre-trained on single chain sequences, while some of our datasets consist of multi-chain proteins. Therefore, when computing ESM-2 embeddings or fine-tuning ESM-1v we proceed as follows. For each sequence we: i) obtain the chain boundaries from the associated reference structure; ii) compute inde...
-
[12]
for similar observations. By contrast BLOSUM50-based zero-shot scores are consistently positive except for the two thermostability landscapes from Tsuboyama et al. (2023) and some of the antibody landscapes from Moulana et al. (2023). Swapping ESM2-8M for ESM-650M in ridge regression and MLP models has mixed effects on performance. While Ridge-ESM2-650M o...
arXiv 2023
-
[13]
Metrics are averaged across 21 datasets. For each column the best performing metric is marked in bold. This table is identical to Table 1, except that it contains additional models (marked in purple). We do not include MAE metrics for zero-shot methods, since they are wildly off-scale. Note that Ridge-ESM2-8M is referred to as Ridge-ESM2 in the main text....
arXiv 2007
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.