Site Preferences and "Coloring Problem" in Cu-doped BiMn₇O₁₂ Quadruple Perovskite
Pith reviewed 2026-06-27 08:40 UTC · model grok-4.3
The pith
Single-crystal refinements place Cu at octahedral Mn sites in lightly doped BiMn7O12, while first-principles calculations place it at square-planar sites.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
In Cu-doped BiMn7O12 (x = 0.05–0.15), single-crystal refinements reveal enhanced electron density at the octahedral B sites of the MnO6 network, indicating preferential Cu occupation there rather than at the expected square-planar A' sites. First-principles calculations instead favor square-planar occupation. The discrepancy points to strong competition among local bonding preferences, short-range disorder, and metastability within the monoclinic I2/m framework that progressively approaches pseudo-cubic symmetry with added copper.
What carries the argument
Site occupancy refinement from single-crystal X-ray diffraction, which maps electron density onto the octahedral Mn B positions versus the square-planar A' positions and is contrasted with DFT total-energy rankings of the two configurations.
If this is right
- The average monoclinic distortion decreases and the lattice metric approaches cubic symmetry with rising Cu content.
- Local structural coherence drops and medium-range order weakens even while the long-range quadruple-perovskite framework persists.
- Two magnetic anomalies near 100–120 K and 50–60 K are progressively suppressed, together with reduced field-induced magnetization.
- Metastability and short-range disorder become dominant factors controlling site choice in this highly frustrated system.
Where Pith is reading between the lines
- If octahedral Cu occupation is real, the usual crystal-chemical rules for Cu2+ square-planar preference may need revision when strong geometric frustration is present.
- The observed competition suggests that synthesis conditions or slight off-stoichiometry could be used to toggle between the two site preferences in related quadruple perovskites.
- Magnetic irreversibility and unsaturated moments may arise from the same local disorder that affects site occupancy, linking structural and magnetic degrees of freedom more tightly than average-structure models imply.
Load-bearing premise
That the extra electron density seen in the refinements comes from copper atoms rather than from vacancies, defects, or the increasing local disorder already noted in the PDF analysis.
What would settle it
A direct local probe such as Cu K-edge EXAFS or element-specific STEM mapping that either confirms or rules out significant copper occupation on the octahedral B sites.
Figures
read the original abstract
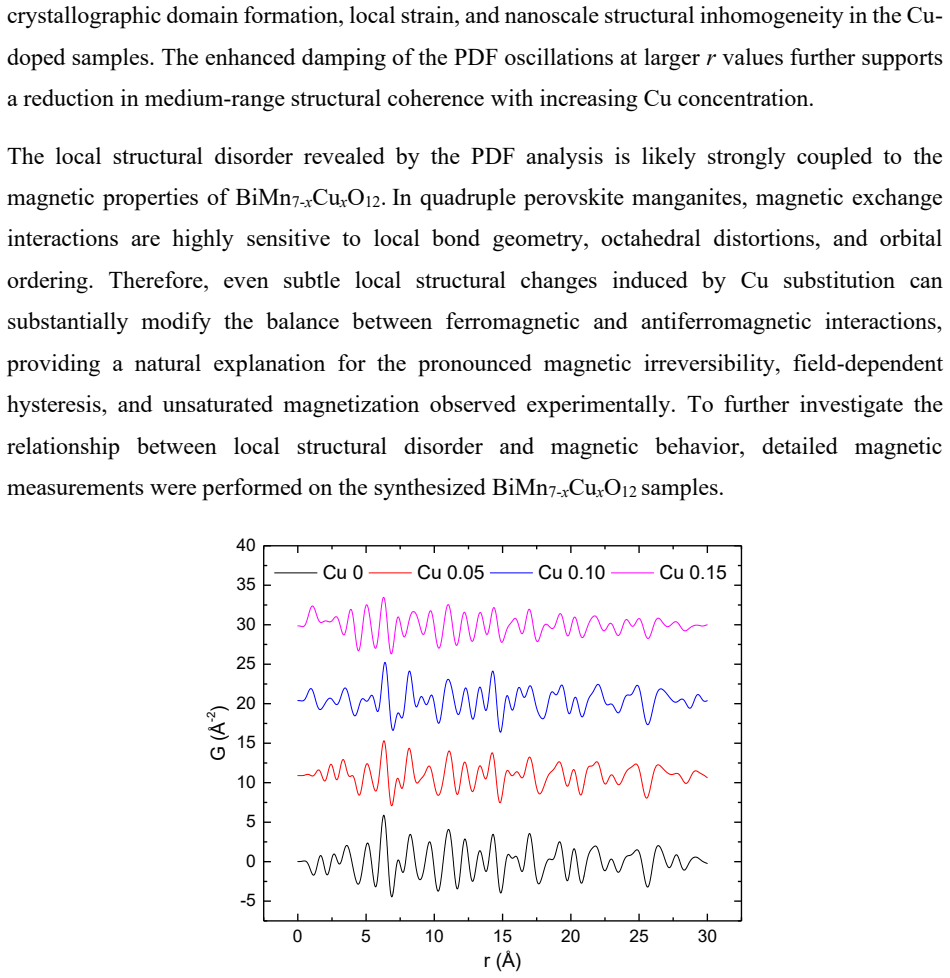
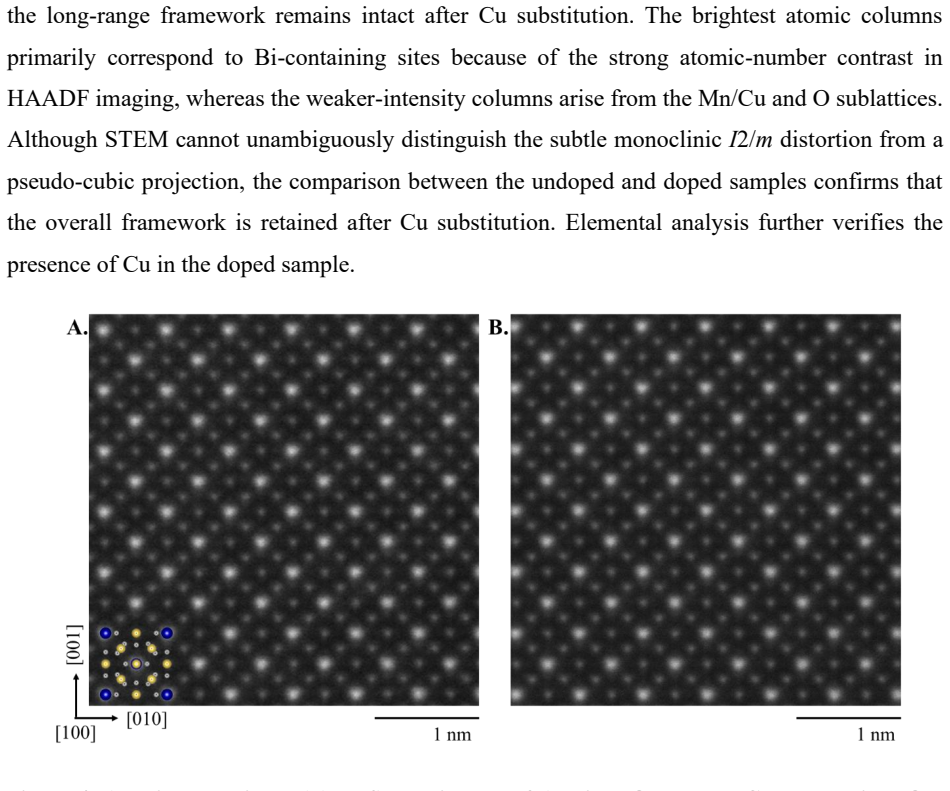
Lightly Cu-doped BiMn$_7$O$_{12}$ (x = 0.05, 0.10, and 0.15) was investigated using high-pressure synthesis, single-crystal X-ray diffraction, pair distribution function (PDF) analysis, STEM, magnetic measurements, and first-principles calculations. All compositions retain an average monoclinic $I$2/$m$ structure, while Cu substitution progressively suppresses the monoclinic distortion and drives the lattice toward a pseudo-cubic metric symmetry. PDF analysis reveals increasing local structural disorder and reduced medium-range coherence with increasing Cu concentration, despite preservation of the overall quadruple-perovskite framework. Single-crystal refinements indicate enhanced electron density at the octahedral Mn B sites, suggesting preferential Cu occupation within the MnO$_6$ network rather than the conventional square-planar sites expected for Cu$^{2+}$. Magnetic measurements reveal two characteristic anomalies near $T_1$ ~ 100-120 K and $T_2$ ~ 50-60 K, together with pronounced magnetic irreversibility, field-dependent hysteresis, and unsaturated magnetization. Increasing Cu concentration progressively suppresses the low-temperature magnetic state and weakens the field-induced moment. First-principles calculations favor Cu occupation at the square-planar sites, contrasting with the experimental refinements and highlighting strong competitions among local bonding, short-range disorder, and metastability in this highly frustrated quadruple perovskite system.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
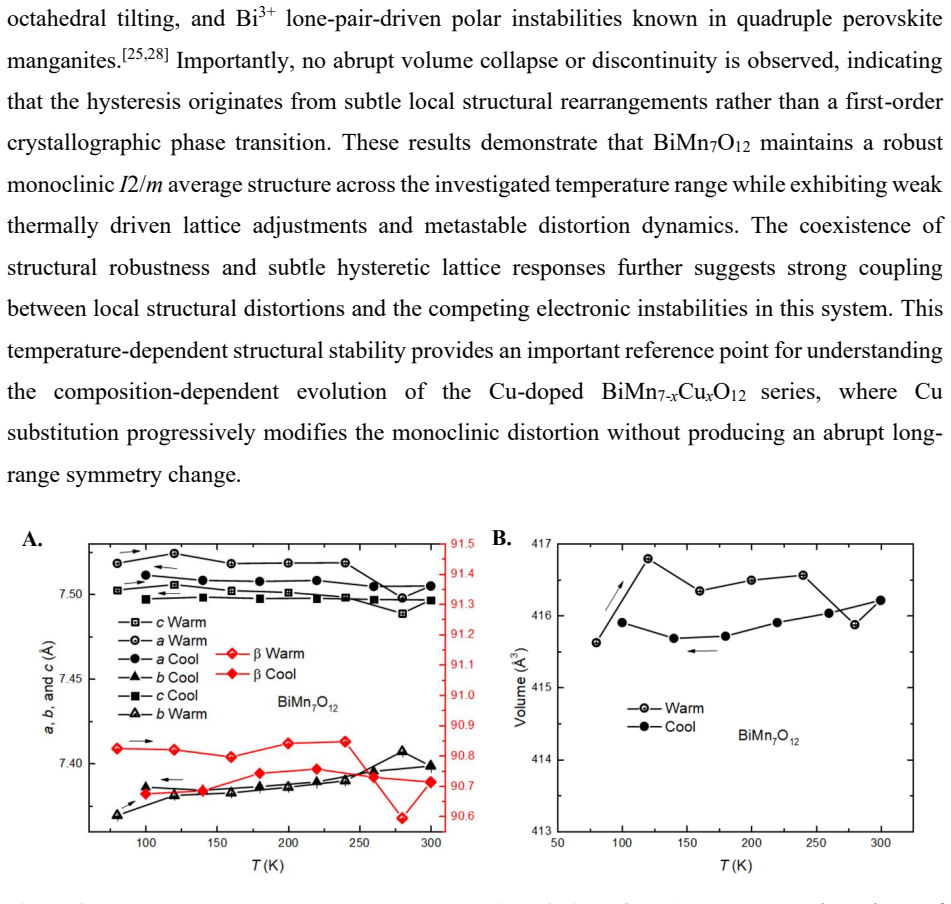
Summary. The manuscript examines lightly Cu-doped BiMn7O12 (x = 0.05, 0.10, 0.15) synthesized under high pressure. Single-crystal X-ray diffraction refinements, PDF analysis, STEM, magnetic measurements, and first-principles calculations are used to show retention of the average I2/m monoclinic structure with progressive suppression of the monoclinic distortion toward pseudo-cubic symmetry. PDF reveals increasing local disorder and loss of medium-range coherence. Single-crystal refinements report enhanced electron density at the octahedral Mn B sites, interpreted as preferential Cu occupation in the MnO6 network rather than conventional square-planar A' sites. DFT calculations instead favor square-planar occupation. Magnetic data show anomalies near 100-120 K and 50-60 K that are suppressed with increasing Cu, along with irreversibility and unsaturated magnetization.
Significance. If the reported experimental-theoretical contrast is robust, the work illustrates competitions among local bonding preferences, short-range disorder, and metastability in a highly frustrated quadruple perovskite, with potential implications for understanding site selectivity and magnetic behavior in related doped systems.
major comments (1)
- [single-crystal refinements and PDF analysis] The central experimental claim (enhanced electron density at octahedral Mn B sites indicating Cu preference over square-planar A' sites) rests on single-crystal refinements whose interpretation is not shown to be robust against the increasing local structural disorder reported in the PDF analysis. Alternative contributions to the observed density (e.g., vacancies, partial Mn displacements, or disorder-induced smearing) are not explicitly tested or quantified, yet this mapping is load-bearing for the headline contrast with the DFT results favoring square-planar sites.
Simulated Author's Rebuttal
We thank the referee for their careful reading of the manuscript and for identifying a key point regarding the robustness of our experimental interpretation. We address the major comment below and outline revisions to strengthen the presentation.
read point-by-point responses
-
Referee: The central experimental claim (enhanced electron density at octahedral Mn B sites indicating Cu preference over square-planar A' sites) rests on single-crystal refinements whose interpretation is not shown to be robust against the increasing local structural disorder reported in the PDF analysis. Alternative contributions to the observed density (e.g., vacancies, partial Mn displacements, or disorder-induced smearing) are not explicitly tested or quantified, yet this mapping is load-bearing for the headline contrast with the DFT results favoring square-planar sites.
Authors: We agree that the PDF results document increasing local disorder with Cu content, which could in principle influence the average-structure refinements. The single-crystal data were refined against the long-range I2/m model, yielding statistically significant electron-density enhancements at the B sites that exceed the nominal Mn scattering and are reproducible across crystals. Vacancies are expected to be minimal under the high-pressure synthesis conditions used, and any Mn displacements are already parameterized via the refined anisotropic displacement tensors. Nevertheless, we acknowledge that explicit model comparisons were not presented. In the revised manuscript we will add a new subsection that (i) reports additional refinements with constrained mixed Mn/Cu occupancies at B sites versus A' sites, (ii) quantifies the effect of introducing artificial smearing or partial vacancies on the refined densities, and (iii) discusses why the observed residuals and bond-valence sums continue to favor the B-site Cu model. These additions will make the experimental–DFT contrast more transparent without altering the central conclusions. revision: yes
Circularity Check
No significant circularity; independent experimental refinements and first-principles calculations
full rationale
The paper derives its central claims from two independent sources: single-crystal X-ray diffraction refinements (yielding electron density maps at Mn B sites) and separate first-principles calculations (favoring square-planar occupation). Neither reduces to a self-defined parameter, a fitted input renamed as prediction, nor a load-bearing self-citation chain. PDF analysis of disorder is reported as an additional observation rather than an input that forces the occupancy interpretation. The reported contrast between experiment and theory is presented as evidence of physical competition, not a tautology. This is the normal case of a self-contained paper with no circular steps.
Axiom & Free-Parameter Ledger
axioms (1)
- domain assumption All compositions retain an average monoclinic I2/m structure
Reference graph
Works this paper leans on
-
[1]
Chenavas, J
J. Chenavas, J. C. Joubert, M. Marezio, B. Bochu, Journal of Solid State Chemistry 1975, 14, 25
1975
-
[2]
M. A. Subramanian, A. W. Sleight, Solid State Sciences 2002, 4, 347
2002
-
[3]
A. N. Vasil’ev, O. S. Volkova, Low Temp. Phys. 2007, 33, 895
2007
-
[4]
L. Zhou, J. Dai, Y. Chai, H. Zhang, S. Dong, H. Cao, S. Calder, Y. Yin, X. Wang, X. Shen, Z. Liu, T. Saito, Y. Shimakawa, H. Hojo, Y. Ikuhara, M. Azuma, Z. Hu, Y. Sun, C. Jin, Y. Long, Advanced Materials 2017, 29, 1703435
2017
-
[5]
Yamada, Science and Technology of Advanced Materials 2017, 18, 541
I. Yamada, Science and Technology of Advanced Materials 2017, 18, 541
2017
-
[6]
Bochu, J
B. Bochu, J. Chenavas, J. C. Joubert, M. Marezio, Journal of Solid State Chemistry 1974, 11, 88
1974
-
[7]
Gilioli, F
E. Gilioli, F. Licci, G. Calestani, A. Prodi, A. Gauzzi, G. Salviati, Crystal Research and Technology 2005, 40, 1072
2005
-
[8]
Okamoto, M
H. Okamoto, M. Karppinen, H. Yamauchi, H. Fjellvåg, Solid State Sciences 2009, 11, 1211
2009
-
[9]
Mezzadri, M
F. Mezzadri, M. Calicchio, E. Gilioli, R. Cabassi, F. Bolzoni, G. Calestani, F. Bissoli, Phys. Rev. B 2009, 79, 014420
2009
-
[10]
Prodi, E
A. Prodi, E. Gilioli, R. Cabassi, F. Bolzoni, F. Licci, Q. Huang, J. W. Lynn, M. Affronte, A. Gauzzi, M. Marezio, Phys. Rev. B 2009, 79, 085105
2009
-
[11]
Cabassi, F
R. Cabassi, F. Bolzoni, E. Gilioli, F. Bissoli, A. Prodi, A. Gauzzi, Phys. Rev. B 2010, 81, 214412
2010
-
[12]
Verseils, F
M. Verseils, F. Mezzadri, D. Delmonte, B. Baptiste, Y. Klein, S. Shcheka, L. C. Chapon, T. Hansen, E. Gilioli, A. Gauzzi, Phys. Rev. Mater. 2017, 1, 064407
2017
-
[13]
Zhang, N
L. Zhang, N. Terada, R. D. Johnson, D. D. Khalyavin, P. Manuel, Y. Katsuya, M. Tanaka, Y. Matsushita, K. Yamaura, A. A. Belik, Inorg. Chem. 2018, 57, 5987
2018
-
[14]
Sławiński, R
W. Sławiński, R. Przeniosło, I. Sosnowska, M. Bieringer, I. Margiolaki, E. Suard, Acta Cryst B 2009, 65, 535
2009
-
[15]
Zhang, S
G. Zhang, S. Dong, Z. Yan, Y. Guo, Q. Zhang, S. Yunoki, E. Dagotto, J.-M. Liu, Phys. Rev. B 2011, 84, 174413
2011
-
[16]
R. D. Johnson, L. C. Chapon, D. D. Khalyavin, P. Manuel, P. G. Radaelli, C. Martin, Phys. Rev. Lett. 2012, 108, 067201
2012
-
[17]
N. J. Perks, R. D. Johnson, C. Martin, L. C. Chapon, P. G. Radaelli, Nat Commun 2012, 3, 1277
2012
-
[18]
Imamura, M
N. Imamura, M. Karppinen, T. Motohashi, D. Fu, M. Itoh, H. Yamauchi, J. Am. Chem. Soc. 2008, 130, 14948
2008
-
[19]
Mezzadri, G
F. Mezzadri, G. Calestani, M. Calicchio, E. Gilioli, F. Bolzoni, R. Cabassi, M. Marezio, A. Migliori, Phys. Rev. B 2009, 79, 100106
2009
-
[20]
Okamoto, N
H. Okamoto, N. Imamura, M. Karppinen, H. Yamauchi, H. Fjellvåg, Inorg. Chem. 2010, 49, 8709
2010
-
[21]
Okamoto, N
H. Okamoto, N. Imamura, M. Karppinen, H. Yamauchi, H. Fjellvåg, Journal of Solid State Chemistry 2010, 183, 186
2010
-
[22]
Imamura, K
N. Imamura, K. Singh, D. Pelloquin, Ch. Simon, T. Sasagawa, M. Karppinen, H. Yamauchi, A. Maignan, Appl. Phys. Lett. 2011, 98, 072903
2011
-
[23]
Gauzzi, G
A. Gauzzi, G. Rousse, F. Mezzadri, G. L. Calestani, G. André, F. Bourée, M. Calicchio, E. Gilioli, R. Cabassi, F. Bolzoni, A. Prodi, P. Bordet, M. Marezio, J. Appl. Phys. 2013, 113, 043920
2013
-
[24]
W. A. Sławiński, H. Okamoto, H. Fjellvåg, Acta Cryst B 2017, 73, 313
2017
-
[25]
A. A. Belik, Y. Matsushita, Y. Kumagai, Y. Katsuya, M. Tanaka, S. Yu. Stefanovich, B. I. Lazoryak, F. Oba, K. Yamaura, Inorg. Chem. 2017, 56, 12272
2017
-
[26]
D. Behr, A. A. Belik, D. D. Khalyavin, R. D. Johnson, Phys. Rev. B 2023, 107, L140402
2023
-
[27]
A. Maia, M. Kempa, V. Bovtun, R. Vilarinho, C. Kadlec, J. Agostinho Moreira, A. A. Belik, P. Proschek, S. Kamba, Phys. Rev. B 2024, 109, 134111
2024
-
[28]
I. S. Soboleva, V. I. Nitsenko, A. V. Sobolev, M. N. Smirnova, A. A. Belik, I. A. Presniakov, International Journal of Molecular Sciences 2024, 25, 1437
2024
-
[29]
D. D. Khalyavin, R. D. Johnson, F. Orlandi, P. G. Radaelli, P. Manuel, A. A. Belik, Science 2020, 369, 680
2020
-
[30]
A. A. Belik, Y. Matsushita, M. Tanaka, R. D. Johnson, D. D. Khalyavin, Journal of Materials Chemistry C 2021, 9, 10232
2021
-
[31]
Li, in Static and Dynamic High Pressure Mineral Physics (Eds.: M
J. Li, in Static and Dynamic High Pressure Mineral Physics (Eds.: M. J. Walter, Y. Fei), Cambridge University Press, Cambridge, 2022, pp. 266–299
2022
-
[32]
Walker, J
D. Walker, J. Li, Matter and Radiation at Extremes 2020, 5, 018402
2020
-
[33]
Parkin, B
S. Parkin, B. Moezzi, H. Hope, J Appl Crystallogr 1995, 28, 53
1995
-
[34]
Walker, D
N. Walker, D. Stuart, Acta Crystallogr A Found Crystallogr 1983, 39, 158
1983
-
[35]
G. M. Sheldrick, Acta Cryst A 2015, 71, 3
2015
-
[36]
G. M. Sheldrick, Acta Cryst C 2015, 71, 3
2015
-
[37]
C. L. Farrow, P. Juhas, J. W. Liu, D. Bryndin, E. S. Božin, J. Bloch, T. Proffen, S. J. L. Billinge, J. Phys.: Condens. Matter 2007, 19, 335219
2007
-
[38]
Juhás, T
P. Juhás, T. Davis, C. L. Farrow, S. J. L. Billinge, J Appl Cryst 2013, 46, 560
2013
-
[39]
Giannozzi, S
P. Giannozzi, S. Baroni, N. Bonini, M. Calandra, R. Car, C. Cavazzoni, D. Ceresoli, G. L. Chiarotti, M. Cococcioni, I. Dabo, A. Dal Corso, S. de Gironcoli, S. Fabris, G. Fratesi, R. Gebauer, U. Gerstmann, C. Gougoussis, A. Kokalj, M. Lazzeri, L. Martin-Samos, N. Marzari, F. Mauri, R. Mazzarello, S. Paolini, A. Pasquarello, L. Paulatto, C. Sbraccia, S. Sca...
2009
-
[40]
Giannozzi, O
P. Giannozzi, O. Andreussi, T. Brumme, O. Bunau, M. Buongiorno Nardelli, M. Calandra, R. Car, C. Cavazzoni, D. Ceresoli, M. Cococcioni, N. Colonna, I. Carnimeo, A. Dal Corso, S. de Gironcoli, P. Delugas, R. A. DiStasio, A. Ferretti, A. Floris, G. Fratesi, G. Fugallo, R. Gebauer, U. Gerstmann, F. Giustino, T. Gorni, J. Jia, M. Kawamura, H.-Y. Ko, A. Kokalj...
2017
-
[41]
Dal Corso, Computational Materials Science 2014, 95, 337
A. Dal Corso, Computational Materials Science 2014, 95, 337
2014
-
[42]
J. P. Perdew, K. Burke, M. Ernzerhof, Phys. Rev. Lett. 1996, 77, 3865
1996
-
[43]
H. J. Monkhorst, J. D. Pack, Phys. Rev. B 1976, 13, 5188
1976
-
[44]
E. R. Davidson, Journal of Computational Physics 1975, 17, 87
1975
-
[45]
Coloring Problem
A. A. Belik, Y. Matsushita, D. D. Khalyavin, Angewandte Chemie International Edition 2017, 56, 10423. Supplementary Information Site Preferences and “Coloring Problem” in Cu-doped BiMn7O12 Quadruple Perovskite Cheng Peng1&, Mingyu Xu1&, Yang Zhang2, Ismail El Baggari2,3, Jie Li4, Weiwei Xie1*
2017
-
[46]
Department of Chemistry, Michigan State University, East Lansing, MI 48824 USA
-
[47]
The Rowland Institute at Harvard, Harvard University, Cambridge, MA 02138 USA
-
[48]
Department of Physics, University of British Columbia, Vancouver, BC V6T 1Z4 Canada
-
[49]
Weiwei Xie (xieweiwe@msu.edu) &equally contributed Table of Contents Table S1
Department of Earth and Environmental Sciences, University of Michigan, Ann Arbor, MI 48109 USA *Corresponding author: Dr. Weiwei Xie (xieweiwe@msu.edu) &equally contributed Table of Contents Table S1. Total scattering structure function 𝐹(𝑄) and corresponding pair distribution function 𝐺(𝑟) of BiMn7O12........................................................
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.