Intrinsic Ductility from Shear Amorphization: From Pure Metals to Multi-Principal-Element Alloys
Pith reviewed 2026-06-27 08:38 UTC · model grok-4.3
The pith
Activation energy density for amorphization predicts intrinsic ductility in metals and alloys from pure elements to multi-principal systems.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
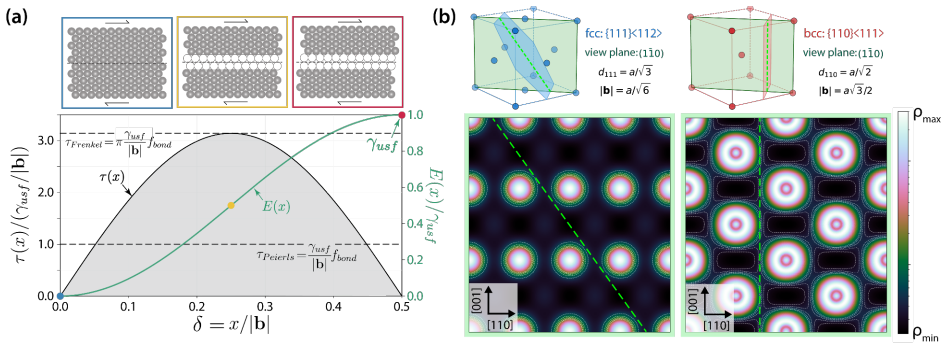
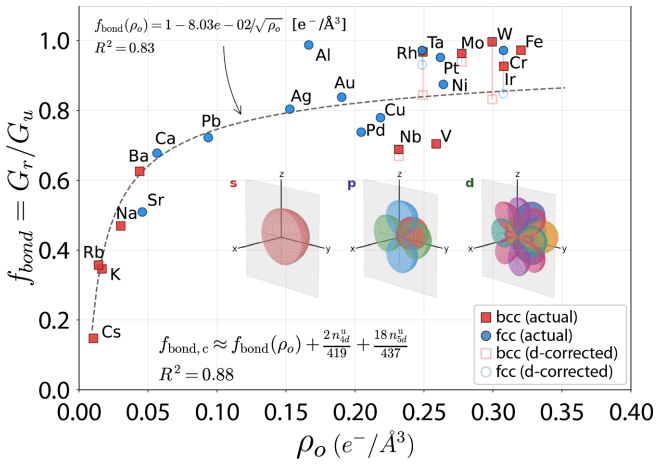
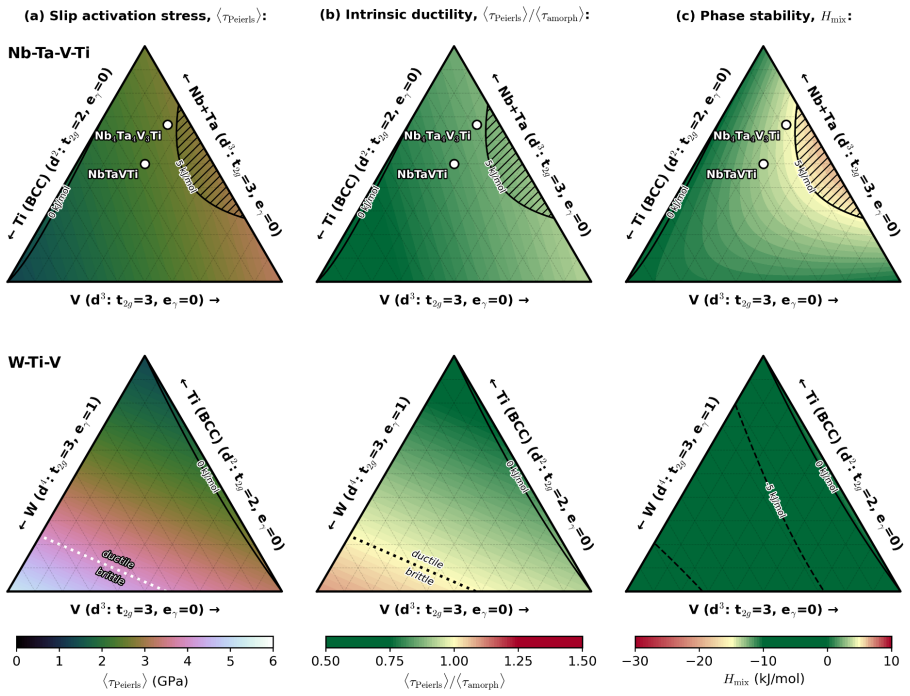
Rather than relying on crystal cleavage and dislocation nucleation at preexisting crack tips, the activation energy density for amorphization serves as a lower energy fracture criterion that enables accurate predictions of both intrinsic ductility and ductile-to-brittle transition temperatures. Analytical expressions combined with ab-initio data yield a unified theory reconciling ductile flow in pure metals and solid-solution alloys, with phase diagrams for the Nb-Ta-V-Ti system explaining its high strength and room-temperature tensile ductility.
What carries the argument
The activation energy density for amorphization, which acts as the controlling lower-energy fracture criterion at crack tips.
If this is right
- Predictions of ductility can be made directly from tabulated ab-initio stiffness constants, lattice parameters, and binary interaction energies.
- The same framework applies uniformly to pure metals and multi-principal-element alloys.
- Phase diagrams can simultaneously account for strength and ductility in alloy systems like Nb-Ta-V-Ti.
- The approach supports rapid computational design of new structural alloys.
Where Pith is reading between the lines
- The reliance on binary interaction energies suggests the model could extend to complex alloys by approximating higher-order terms.
- Experimental observation of amorphization at crack tips before cleavage in ductile samples would provide direct support.
- Linking the criterion more explicitly to electronic band filling could refine predictions for specific elemental combinations.
Load-bearing premise
Activation energy density for amorphization is lower than the energy for crystal cleavage and dislocation nucleation at preexisting crack tips, making it the governing factor for intrinsic ductility.
What would settle it
Finding a ductile metal where the calculated activation energy density for amorphization exceeds the energy barriers for cleavage or dislocation nucleation at crack tips.
Figures




read the original abstract
Direct links between electronic structure and intrinsic ductility remain elusive for metals. A framework is proposed that reduces the complexities of valence charge distribution, band filling, and shear strain effects into structure-property relationships describing the intrinsic ductility of metals and alloys. Rather than relying on crystal cleavage and dislocation nucleation at preexisting crack tips, we show that a lower energy fracture criterion, i.e., the activation energy density for amorphization, enables accurate predictions of both intrinsic ductility and ductile-to-brittle transition temperatures. From analytical expressions and tabulated ab-initio stiffness constants, lattice parameters, and binary interaction energies, we present a unified theory that reconciles ductile flow in pure metals and solid-solution alloys. Phase diagrams generated for the Nb-Ta-V-Ti system simultaneously explain its high strength and room-temperature tensile ductility, validating this framework as a practical one for rapid design of structural multi-principal-element alloys.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript proposes a unified theoretical framework for intrinsic ductility and ductile-to-brittle transition temperatures in pure metals and multi-principal-element alloys. It asserts that the activation energy density for shear amorphization constitutes a lower-energy fracture criterion than crystal cleavage or dislocation nucleation at preexisting crack tips. Analytical expressions derived from tabulated ab-initio stiffness constants, lattice parameters, and binary interaction energies are used to generate predictions, with validation claimed via phase diagrams for the Nb-Ta-V-Ti system that account for its observed high strength and room-temperature tensile ductility.
Significance. If the central claims hold, the work would represent a meaningful advance by supplying an analytical, ab-initio-based route to ductility predictions that spans pure metals and complex solid-solution alloys, with direct utility for rapid MPEA design. The reliance on tabulated parameters rather than extensive new computations is a constructive feature that could facilitate broader application.
major comments (3)
- [Abstract and fracture-criterion derivation] Abstract and the section deriving the fracture criterion: The manuscript states that amorphization activation energy density is a lower-energy criterion enabling the ductility predictions, yet does not furnish an explicit numerical comparison or inequality (E_amorph < min(E_cleavage, E_disloc)) evaluated at crack tips for the modeled systems. This demonstration is load-bearing for the claim that amorphization governs intrinsic ductility rather than serving as an alternative parameterization.
- [Nb-Ta-V-Ti validation] Validation section on Nb-Ta-V-Ti: The phase diagrams are presented as simultaneously explaining strength and ductility, but the text provides no tabulated quantitative metrics (e.g., predicted versus measured ductility values, DBTT errors, or R² statistics) that would allow independent assessment of the asserted accuracy of the predictions.
- [Methods / input parameters] Methods section on input parameters: Binary interaction energies are drawn from tabulated ab-initio sources; if any of these energies were fitted to mechanical-property data overlapping with the ductility or DBTT validation sets, the framework would contain a circularity that undermines the claim of a new governing criterion derived from first principles.
minor comments (1)
- All symbols appearing in the analytical expressions for activation energy density should be defined at first use, including any auxiliary quantities derived from stiffness constants and lattice parameters.
Simulated Author's Rebuttal
We thank the referee for the constructive comments, which help improve the clarity and rigor of our manuscript. We respond to each major comment point by point below.
read point-by-point responses
-
Referee: [Abstract and fracture-criterion derivation] Abstract and the section deriving the fracture criterion: The manuscript states that amorphization activation energy density is a lower-energy criterion enabling the ductility predictions, yet does not furnish an explicit numerical comparison or inequality (E_amorph < min(E_cleavage, E_disloc)) evaluated at crack tips for the modeled systems. This demonstration is load-bearing for the claim that amorphization governs intrinsic ductility rather than serving as an alternative parameterization.
Authors: We agree that an explicit numerical comparison would strengthen the central claim. In the revised manuscript we will add a dedicated subsection with tabulated values of E_amorph, E_cleavage and E_disloc evaluated at crack tips for representative pure metals and Nb-Ta-V-Ti compositions, confirming the inequality using the same ab-initio stiffness and interaction parameters already employed in the framework. revision: yes
-
Referee: [Nb-Ta-V-Ti validation] Validation section on Nb-Ta-V-Ti: The phase diagrams are presented as simultaneously explaining strength and ductility, but the text provides no tabulated quantitative metrics (e.g., predicted versus measured ductility values, DBTT errors, or R² statistics) that would allow independent assessment of the asserted accuracy of the predictions.
Authors: We acknowledge the absence of quantitative metrics. The revised manuscript will include a new table listing predicted versus experimentally reported ductility indicators and DBTT values for the Nb-Ta-V-Ti alloys, together with mean absolute errors and any available correlation statistics to permit independent assessment. revision: yes
-
Referee: [Methods / input parameters] Methods section on input parameters: Binary interaction energies are drawn from tabulated ab-initio sources; if any of these energies were fitted to mechanical-property data overlapping with the ductility or DBTT validation sets, the framework would contain a circularity that undermines the claim of a new governing criterion derived from first principles.
Authors: The binary interaction energies are taken exclusively from published ab-initio DFT studies of thermodynamic mixing enthalpies that predate and are independent of the mechanical-property validation sets used here. No fitting to ductility or DBTT data occurred, so the framework contains no circularity. revision: no
Circularity Check
No significant circularity; derivation uses independent tabulated ab-initio inputs
full rationale
The abstract presents analytical expressions derived from tabulated ab-initio stiffness constants, lattice parameters, and binary interaction energies as external inputs to generate predictions for ductility and DBTT. No equations or steps are shown that reduce the activation energy density for amorphization to a fitted parameter or self-citation by construction. The claim that amorphization is a lower-energy criterion is presented as a derived result from those inputs rather than an assumption smuggled in via self-reference. This is a standard use of external computational data and does not trigger any of the enumerated circularity patterns.
Axiom & Free-Parameter Ledger
free parameters (1)
- binary interaction energies
axioms (1)
- domain assumption Shear amorphization activation energy density is the controlling fracture criterion for intrinsic ductility
Reference graph
Works this paper leans on
-
[1]
Hall, E. O. The Deformation and Ageing of Mild Steel: III Discussion of Results. Proc. Phys. Soc. Sect. B 64, 747–753 (1951)
1951
-
[2]
Petch, N. J. The Cleavage Strength of Polycrystals. J. Iron Steel Inst. 173, 25–28 (1953)
1953
-
[3]
C., Knight, B
Cordero, Z. C., Knight, B. E. & Schuh, C. A. Six decades of the Hall–Petch effect – a survey of grain-size strengthening studies on pure metals. Int. Mater. Rev. 61, 495–512 (2016)
2016
-
[4]
Taylor, G. I. The mechanism of plastic deformation of crystals. Part I.—Theoretical. Proc. R. Soc. Lond. Ser. Contain. Pap. Math. Phys. Character 145, 362–387 (1934)
1934
-
[5]
Taylor, G. I. The Mechanism of Plastic Deformation of Crystals. Part II. Comparison with Observations. Proc. R. Soc. Math. Phys. Eng. Sci. 145, 388–404 (1934)
1934
-
[6]
Zur Kristallplastizität
Orowan, E. Zur Kristallplastizität. I. Z. Für Phys. 89, 605–613 (1934)
1934
-
[7]
Fleischer, R. L. Solution hardening. Acta Metall. 9, 996–1000 (1961)
1961
-
[8]
Fleischer, R. L. Substitutional solution hardening. Acta Metall. 11, 203–209 (1963)
1963
-
[9]
Cottrell, A. H. & Bilby, B. A. Dislocation Theory of Yielding and Strain Ageing of Iron. Proc. Phys. Soc. Sect. A 62, 49 (1949)
1949
-
[10]
A Statistical Theory of Solid Solution Hardening
Labusch, R. A Statistical Theory of Solid Solution Hardening. Phys. Status Solidi B 41, 659– 669 (1970)
1970
-
[11]
Statistische theorien der mischkristallhärtung
Labusch, R. Statistische theorien der mischkristallhärtung. Acta Metall. 20, 917–927 (1972)
1972
-
[12]
Mott, N. F. & Nabarro, F. R. N. An attempt to estimate the degree of precipitation hardening, with a simple model. Proc. Phys. Soc. 52, 86 (1940)
1940
-
[13]
J., Kocks, U
Bacon, D. J., Kocks, U. F. & Scattergood, R. O. The effect of dislocation self-interaction on the orowan stress. Philos. Mag. J. Theor. Exp. Appl. Phys. 28, 1241–1263 (1973)
1973
-
[14]
Ashby, M. F. The deformation of plastically non-homogeneous materials. Philos. Mag. J. Theor. Exp. Appl. Phys. 21, 399–424 (1970)
1970
-
[15]
Chookajorn, T., Murdoch, H. A. & Schuh, C. A. Design of Stable Nanocrystalline Alloys. Science 337, 951–954 (2012)
2012
-
[16]
Murdoch, H. A. & Schuh, C. A. Stability of binary nanocrystalline alloys against grain growth and phase separation. Acta Mater. 61, 2121–2132 (2013)
2013
-
[17]
Pugh, S. F. XCII. Relations between the elastic moduli and the plastic properties of polycrystalline pure metals. Lond. Edinb. Dublin Philos. Mag. J. Sci. 45, 823–843 (1954)
1954
-
[18]
N., Greer, A
Greaves, G. N., Greer, A. L., Lakes, R. S. & Rouxel, T. Poisson’s ratio and modern materials. Nat. Mater. 10, 823–837 (2011). 19
2011
-
[19]
Pettifor, D. G. New many-body potential for the bond order. Phys. Rev. Lett. 63, 2480–2483 (1989)
1989
-
[20]
Pettifor, D. G. Theoretical predictions of structure and related properties of intermetallics. Mater. Sci. Technol. 8, 345–349 (1992)
1992
-
[21]
Rice, J. R. Dislocation nucleation from a crack tip: An analysis based on the Peierls concept. J. Mech. Phys. Solids 40, 239–271 (1992)
1992
-
[22]
Irwin, G. R. Analysis of stresses and strains near the end of a crack traversing a plate. J. Appl. Mech. 24, 361–364 (1957)
1957
-
[23]
Griffith, A. A. VI. The phenomena of rupture and flow in solids. Philos. Trans. R. Soc. Lond. Ser. Contain. Pap. Math. Phys. Character 221, 163–198 (1921)
1921
-
[24]
K., Varga, L
Temesi, O. K., Varga, L. K., Chinh, N. Q. & Vitos, L. Ductility Index for Refractory High Entropy Alloys. Crystals 14, (2024)
2024
-
[25]
Singh, P. et al. A ductility metric for refractory-based multi-principal-element alloys. Acta Mater. 257, 119104 (2023)
2023
-
[26]
& Chrzan, D
Qi, L. & Chrzan, D. C. Tuning ideal tensile strengths and intrinsic ductility of bcc refractory alloys. Phys. Rev. Lett. 112, 1–5 (2014)
2014
-
[27]
Hu, Y .-J., Sundar, A., Ogata, S. & Qi, L. Screening of generalized stacking fault energies, surface energies and intrinsic ductile potency of refractory multicomponent alloys. Acta Mater. 210, 116800 (2021)
2021
-
[28]
M., Murty, B
Shaikh, S. M., Murty, B. S. & Yadav, S. K. Designing a thermodynamically stable and intrinsically ductile refractory alloy. J. Alloys Compd. 939, 168597 (2023)
2023
-
[29]
& Gao, M
Raghuraman, V ., Widom, M., San, S. & Gao, M. C. Ab initio tensile tests applied to bcc refractory alloys. Phys. Rev. Mater. 7, 123601 (2023)
2023
-
[30]
Borges, P. P. P. O., Ritchie, R. O. & Asta, M. Electronic descriptors for dislocation deformation behavior and intrinsic ductility in bcc high-entropy alloys. Sci. Adv. 10, eadp7670 (2024)
2024
-
[31]
& Aidhy, D
Pant, D. & Aidhy, D. S. Electronic density of states as the descriptor of elastic bond strength, ductility, and local lattice distortion in BCC refractory alloys. Mater. Des. 253, 113885 (2025)
2025
-
[32]
P., Raabe, D
George, E. P., Raabe, D. & Ritchie, R. O. High-entropy alloys. Nat. Rev. Mater. 4, 515–534 (2019)
2019
-
[33]
Pollock, T. M. Alloy design for aircraft engines. Nat. Mater. 15, 809–815 (2016)
2016
-
[34]
Perepezko, J. H. The Hotter the Engine, the Better. Science 326, 1068–1069 (2009)
2009
-
[35]
Cantor, B., Chang, I. T. H., Knight, P. & Vincent, A. J. B. Microstructural development in equiatomic multicomponent alloys. Mater. Sci. Eng. A 375–377, 213–218 (2004)
2004
-
[36]
Yeh, J. W. et al. Nanostructured high-entropy alloys with multiple principal elements: Novel alloy design concepts and outcomes. Adv. Eng. Mater. 6, 299-303+274 (2004)
2004
-
[37]
Huang, H. et al. Achieving high tensile strength and ductility in refractory alloys by tuning electronic structure. Nat. Mater. 25, 386–394 (2026)
2026
-
[38]
Gludovatz, B. et al. A fracture-resistant high-entropy alloy for cryogenic applications. Science 345, 1153–1158 (2014). 20
2014
-
[39]
Liu, D. et al. Exceptional fracture toughness of CrCoNi-based medium- and high-entropy alloys at 20 kelvin. Science 378, 978–983 (2022)
2022
-
[40]
Wang, F. et al. Multiplicity of dislocation pathways in a refractory multiprincipal element alloy. Science 370, 95–101 (2020)
2020
-
[41]
N., Wilks, G
Senkov, O. N., Wilks, G. B., Miracle, D. B., Chuang, C. P. & Liaw, P. K. Refractory high- entropy alloys. 18, 1758–1765 (2010)
2010
-
[42]
N., Miracle, D
Senkov, O. N., Miracle, D. B., Chaput, K. J. & Couzinie, J. P. Development and exploration of refractory high entropy alloys - A review. J. Mater. Res. 33, 3092–3128 (2018)
2018
-
[43]
Argibay, N. et al. An energetic link between order and strength in metals: A nanocrystalline strength limit in high-entropy alloys and intermetallic compounds. Acta Mater. 290, 120990 (2025)
2025
-
[44]
& Argibay, N
Chandross, M. & Argibay, N. Ultimate Strength of Metals. Phys. Rev. Lett. 124, 125501 (2020)
2020
-
[45]
D., Singh, P., Smirnov, A
Johnson, D. D., Singh, P., Smirnov, A. V . & Argibay, N. Universal Maximum Strength of Solid Metals and Alloys. Phys. Rev. Lett. 130, 166101 (2023)
2023
-
[46]
Chen, W. et al. A map of single-phase high-entropy alloys. Nat. Commun. 14, 2856 (2023)
2023
-
[47]
Shang, S. L. et al. First-principles calculations of pure elements: Equations of state and elastic stiffness constants. Comput. Mater. Sci. 48, 813–826 (2010)
2010
-
[48]
Luo, H., Zhang, H., Sheng, H., Liu, J. P. & Szlufarska, I. Amorphous shear bands in SmCo5. Mater. Sci. Eng. A 785, (2020)
2020
-
[49]
Khrapak, S. A. Lindemann melting criterion in two dimensions. Phys. Rev. Res. 2, 012040 (2020)
2020
-
[50]
L., Walls, H
Hines, A. L., Walls, H. A. & Jethani, K. R. Determination of the coordination number of liquid metals near the melting point. Metall. Trans. A 16, 267–274 (1985)
1985
-
[51]
The Structure of Liquid Transition Metals and Their Alloys
Waseda, Y . The Structure of Liquid Transition Metals and Their Alloys . (Institute of Physics, United Kingdom, 1977)
1977
-
[52]
& Yip, S
Ogata, S., Li, J. & Yip, S. Ideal Pure Shear Strength of Aluminum and Copper. Science 298, 807–811 (2002)
2002
-
[53]
& Yip, S
Ogata, S., Li, J., Hirosaki, N., Shibutani, Y . & Yip, S. Ideal shear strain of metals and ceramics. Phys. Rev. B 70, 104104 (2004)
2004
-
[54]
& Vitos, L
Levämäki, H. & Vitos, L. Electron localization function implementation in the exact muffin- tin orbitals method. Phys. Rev. B 103, 035118 (2021)
2021
-
[55]
& Fässler, T
Savin, A., Nesper, R., Wengert, S. & Fässler, T. F. ELF: The Electron Localization Function. Angew. Chem. Int. Ed. Engl. 36, 1808–1832 (1997)
1997
-
[56]
McAdon, M. H. & Goddard, W. A. New Concepts of Metallic Bonding Based on Valence- Bond Ideas. Phys. Rev. Lett. 55, 2563–2566 (1985)
1985
-
[57]
Xu, S., Su, Y ., Smith, L. T. W. & Beyerlein, I. J. Frank-Read source operation in six body- centered cubic refractory metals. J. Mech. Phys. Solids 141, 104017 (2020)
2020
-
[58]
Cook, D. H. et al. Kink bands promote exceptional fracture resistance in a NbTaTiHf refractory medium-entropy alloy. Science 384, 178–184 (2024). 21
2024
-
[59]
N., Scott, J
Senkov, O. N., Scott, J. M., Senkova, S. V ., Miracle, D. B. & Woodward, C. F. Microstructure and room temperature properties of a high-entropy TaNbHfZrTi alloy. J. Alloys Compd. 509, 6043–6048 (2011)
2011
-
[60]
N., Wilks, G
Senkov, O. N., Wilks, G. B., Scott, J. M. & Miracle, D. B. Mechanical properties of Nb25Mo25Ta 25W25 and V20Nb20Mo 20Ta20W20 refractory high entropy alloys. Intermetallics 19, 698–706 (2011)
2011
-
[61]
Sheikh, S. et al. Alloy design for intrinsically ductile refractory high-entropy alloys. J. Appl. Phys. 120, 164902 (2016)
2016
-
[62]
Yao, H. W. et al. NbTaV-(Ti,W) refractory high-entropy alloys: Experiments and modeling. Mater. Sci. Eng. A 674, 203–211 (2016)
2016
-
[63]
Couzinié, J.-Ph. et al. On the room temperature deformation mechanisms of a TiZrHfNbTa refractory high-entropy alloy. Mater. Sci. Eng. A 645, 255–263 (2015)
2015
-
[64]
Lilensten, L. et al. Study of a bcc multi-principal element alloy: Tensile and simple shear properties and underlying deformation mechanisms. Acta Mater. 142, 131–141 (2018)
2018
-
[65]
Guduru, S. P. R., Kekung, M. O., Ott, R. T., Roy, S. & Singh, P. DuctGPT: A Generative Transformer for Forward Screening of Ductile Refractory Multi-Principal Element Alloys. Acta Mater. 304, 121763 (2026)
2026
-
[66]
& Reiser, J
Bonnekoh, C., Hoffmann, A. & Reiser, J. The brittle-to-ductile transition in cold rolled tungsten: On the decrease of the brittle-to-ductile transition by 600 K to − 65 °C. Int. J. Refract. Met. Hard Mater. 71, 181–189 (2018)
2018
-
[67]
R., Boyce, B
Weinberger, C. R., Boyce, B. L. & Battaile, C. C. Slip planes in bcc transition metals. Int. Mater. Rev. 58, 296–314 (2013)
2013
-
[68]
C., Carroll, J
Lim, H., Battaile, C. C., Carroll, J. D., Boyce, B. L. & Weinberger, C. R. A physically based model of temperature and strain rate dependent yield in BCC metals: Implementation into crystal plasticity. J. Mech. Phys. Solids 74, 80–96 (2014)
2014
-
[69]
Holt, D. L. Dislocation Cell Formation in Metals. J. Appl. Phys. 41, 3197–3201 (1970)
1970
-
[70]
& Rivera-Díaz-del-Castillo, P
Galindo-Nava, E. & Rivera-Díaz-del-Castillo, P. Modelling plastic deformation in BCC metals: Dynamic recovery and cell formation effects. Mater. Sci. Eng. A 558, 641–648 (2012)
2012
-
[71]
Raffo, P. L. Yielding and fracture in tungsten and tungsten-rhenium alloys. J. Common Met. 17, 133–149 (1969)
1969
-
[72]
& Jaffee, R
Schwartzberg, F., Ogden, H. & Jaffee, R. I. Ductile-Brittle Transition in the Refractory Metals. vol. 114 (Defense Metals Information Center, Battelle Memorial Institute, 1959)
1959
-
[73]
Pugh, J. W. Tensile and creep properties of tungsten at elevated temperatures. in Proc. ASTM vol. 57 11 (1957)
1957
-
[74]
S., Frick, C
Schneider, A. S., Frick, C. P., Clark, B. G., Gruber, P. A. & Arzt, E. Influence of orientation on the size effect in bcc pillars with different critical temperatures. Mater. Sci. Eng. A 528, 1540–1547 (2011)
2011
-
[75]
Blöchl, P. E. Projector augmented-wave method. Phys. Rev. B 50, 17953–17979 (1994)
1994
-
[76]
Giannozzi, P. et al. QUANTUM ESPRESSO: a modular and open-source software project for quantum simulations of materials. J. Phys. Condens. Matter 21, 395502 (2009). 22
2009
-
[77]
Giannozzi, P. et al. Advanced capabilities for materials modelling with Quantum ESPRESSO. J. Phys. Condens. Matter 29, 465901 (2017)
2017
-
[78]
P., Burke, K
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys Rev Lett 77, 3865–3868 (1996)
1996
-
[79]
Pseudopotentials periodic table: From H to Pu
Dal Corso, A. Pseudopotentials periodic table: From H to Pu. Comput. Mater. Sci. 95, 337– 350 (2014)
2014
-
[80]
Monkhorst, H. J. & Pack, J. D. Special points for Brillouin-zone integrations. Phys. Rev. B 13, 5188–5192 (1976). 23
1976
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.