Thickness-Dependent Interlayer Coupling and Semiconductor-to-Semimetal Crossover in Arsenene Multilayers
Pith reviewed 2026-06-26 16:46 UTC · model grok-4.3
The pith
Arsenene multilayers change stacking sequence with added layers, collapsing the band gap through stronger pz orbital overlap.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
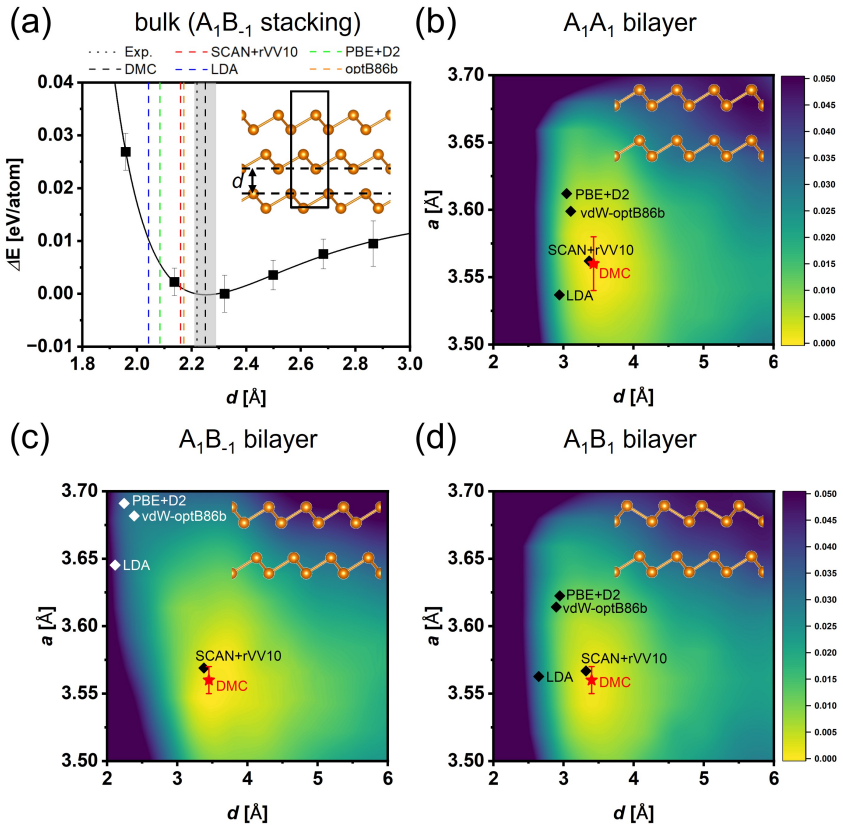
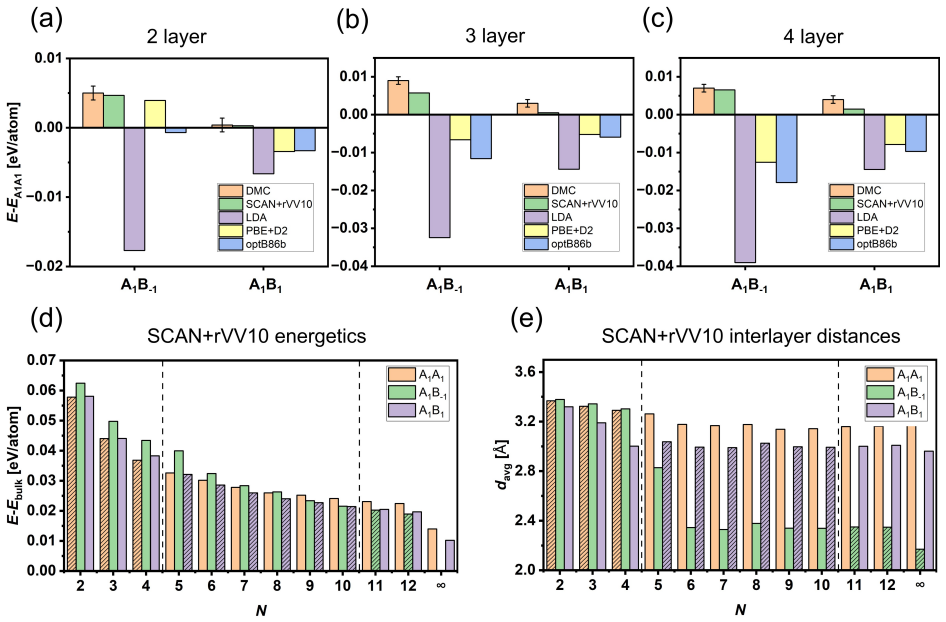
Registry alone does not determine the bonding regime; thickness and coordination reshape the interlayer interaction. DMC establishes that bulk gray arsenic is compact while few-layer structures stay at substantially larger separations despite the same A1B-1 adjacent-layer registry. Among tested functionals, SCAN+rVV10 reproduces the DMC separations and energetics most closely. DMC-benchmarked SCAN+rVV10 calculations predict a thickness-driven stacking sequence from A1A1 to A1B1 and finally bulk-like A1B-1, with the structural crossover coinciding with a stacking-dependent band-gap collapse from enhanced interlayer As pz hybridization.
What carries the argument
Thickness-dependent strengthening of interlayer As pz hybridization that drives both the stacking sequence change and the electronic crossover.
If this is right
- Few-layer arsenene maintains larger interlayer separations than bulk despite sharing the same nominal registry.
- SCAN+rVV10 is the functional whose equilibrium geometries and energies align most closely with DMC benchmarks.
- Stacking evolves with thickness from A1A1 in the thinnest limit through A1B1 to the bulk A1B-1 arrangement.
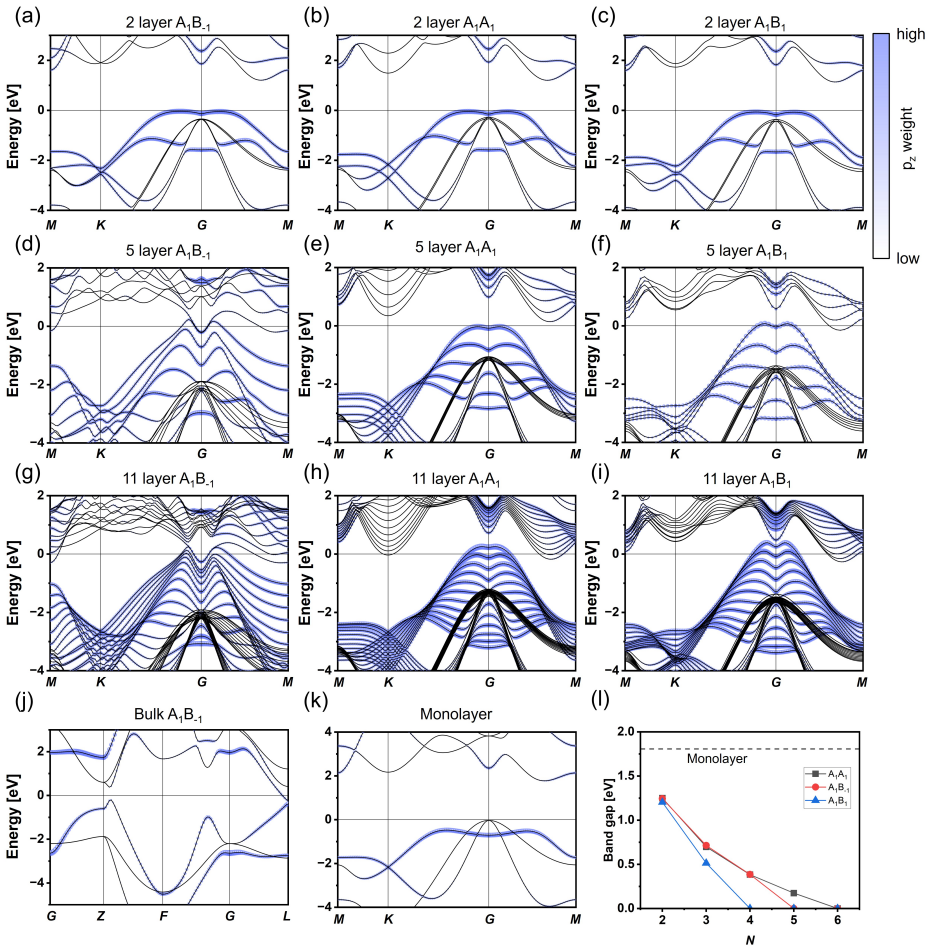
- The stacking change produces a progressive band-gap collapse driven by increasing As pz hybridization.
Where Pith is reading between the lines
- The same thickness-controlled hybridization mechanism could operate in other group-V monolayers whose out-of-plane orbitals participate in bonding.
- Layer-number control might therefore serve as a route to engineer the gap without external fields or strain.
- The crossover point identified here supplies a concrete target thickness for transport or optical experiments that test the predicted semimetal onset.
Load-bearing premise
Diffusion Monte Carlo equilibrium separations and energies provide a reliable benchmark for choosing and validating the SCAN+rVV10 functional at every thickness without significant finite-size or pseudopotential errors.
What would settle it
Experimental measurement of interlayer distance in few-layer arsenene that either matches the larger DMC-predicted separation or collapses to the compact bulk value at the same registry.
Figures
read the original abstract
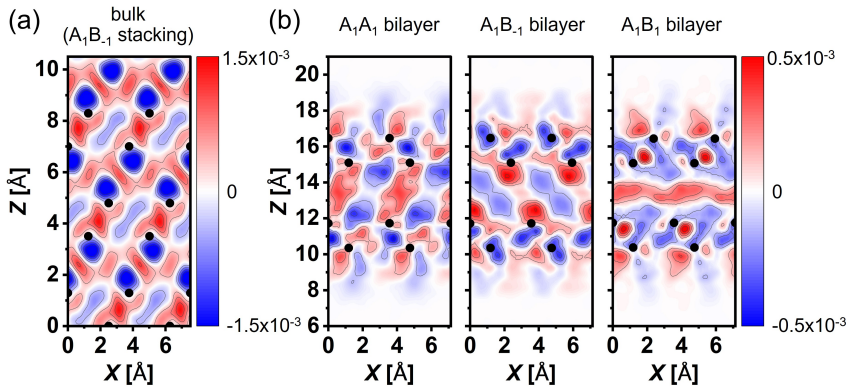
Interlayer interactions in layered materials are often assumed to transfer from the bilayer to the bulk, but this assumption can fail when chemically active out-of-plane orbitals participate in bonding. We combine diffusion quantum Monte Carlo (DMC) and density functional theory (DFT) to determine how interlayer coupling evolves in arsenene multilayers. DMC shows that bulk gray arsenic is compact, whereas the corresponding few-layer structures remain at substantially larger interlayer separations despite sharing the same nominal A$_{1}$B$_{-1}$ adjacent-layer registry. Registry alone therefore does not determine the bonding regime; thickness and coordination reshape the interlayer interaction. Among the tested functionals, SCAN+rVV10 most closely reproduces DMC equilibrium separations and stacking energetics. Using the DMC-benchmarked SCAN+rVV10 calculations, we predict a thickness-driven stacking sequence from A$_{1}$A$_{1}$ to A$_{1}$B$_{1}$ and finally bulk-like A$_{1}$B$_{-1}$. The structural crossover coincides with a stacking-dependent DFT band-gap collapse driven by enhanced interlayer As p$_{z}$ hybridization.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The paper claims that interlayer interactions in arsenene do not transfer uniformly from few-layer to bulk structures when out-of-plane pz orbitals are active. DMC calculations establish that few-layer arsenene remains at larger equilibrium separations than bulk gray arsenic despite identical A1B−1 registry, implying thickness and coordination reshape the bonding regime. Among functionals tested, SCAN+rVV10 best reproduces the DMC separations and energetics; its use then predicts a thickness-driven stacking evolution (A1A1 → A1B1 → bulk-like A1B−1) that coincides with a stacking-dependent band-gap collapse driven by enhanced interlayer As pz hybridization.
Significance. If the DMC benchmark and subsequent DFT predictions hold, the work provides a concrete counter-example to the common assumption that bilayer registry and energetics can be extrapolated to thicker slabs or bulk in materials with chemically active out-of-plane orbitals. The explicit use of DMC as an external benchmark to select and validate SCAN+rVV10, together with the prediction of a semiconductor-to-semimetal crossover tied to stacking change, supplies falsifiable structural and electronic trends that can be tested experimentally.
major comments (2)
- [DMC calculations and benchmark comparison] DMC benchmark section: the central claim that few-layer structures remain at substantially larger separations than bulk (despite shared A1B−1 registry) is used to select SCAN+rVV10 and to justify the thickness-dependent stacking sequence. No explicit finite-size extrapolation data, system-size table, or pseudopotential correction comparison across layer counts (bilayer through bulk) is presented; without these, the reliability of the DMC reference for all thicknesses cannot be assessed and the functional choice becomes circular.
- [DFT structural and electronic results] Results on stacking sequence (§ on DFT structural relaxations): the predicted crossover from A1A1 to A1B1 to A1B−1 is reported as coinciding with the band-gap collapse, yet the energy differences between stackings are not shown with error bars propagated from the DMC benchmark uncertainty. If those differences lie within the DMC statistical or finite-size error, the structural assignment and the attribution of the gap closure to enhanced pz hybridization lose their load-bearing support.
minor comments (2)
- Figure captions and axis labels should explicitly state the number of layers and the functional used for each panel to avoid ambiguity when comparing DMC and DFT curves.
- The abstract states that 'among the tested functionals' SCAN+rVV10 performs best, but the main text should include a compact table listing all functionals examined together with their mean absolute deviations from DMC separations and energies.
Simulated Author's Rebuttal
We thank the referee for the constructive comments on the DMC benchmarks and the robustness of the stacking predictions. We address each major comment point by point below.
read point-by-point responses
-
Referee: [DMC calculations and benchmark comparison] DMC benchmark section: the central claim that few-layer structures remain at substantially larger separations than bulk (despite shared A1B−1 registry) is used to select SCAN+rVV10 and to justify the thickness-dependent stacking sequence. No explicit finite-size extrapolation data, system-size table, or pseudopotential correction comparison across layer counts (bilayer through bulk) is presented; without these, the reliability of the DMC reference for all thicknesses cannot be assessed and the functional choice becomes circular.
Authors: We agree that additional documentation of the DMC technical details is warranted. In the revised manuscript we will add a supplementary table listing the simulation cell sizes (number of atoms and k-point grids) employed for each thickness from bilayer to bulk, together with the finite-size extrapolation procedure and resulting corrections. Pseudopotential locality and core-correction tests were performed during the original calculations and showed variations well below the statistical error across layer counts; we will include a concise summary of these checks in the methods section. These additions will make the DMC reference data transparent and remove any appearance of circularity in the functional selection. revision: yes
-
Referee: [DFT structural and electronic results] Results on stacking sequence (§ on DFT structural relaxations): the predicted crossover from A1A1 to A1B1 to A1B−1 is reported as coinciding with the band-gap collapse, yet the energy differences between stackings are not shown with error bars propagated from the DMC benchmark uncertainty. If those differences lie within the DMC statistical or finite-size error, the structural assignment and the attribution of the gap closure to enhanced pz hybridization lose their load-bearing support.
Authors: The stacking energy differences are a few meV per atom while DMC statistical uncertainties are typically 1–2 meV/atom. In the revision we will propagate these DMC uncertainties as error bars on the stacking-energy plots and tables. We will also add a short paragraph clarifying that the structural sequence is anchored by the DMC-validated SCAN+rVV10 energetics, while the band-gap collapse and pz-hybridization analysis are obtained from the same functional applied to the DMC-preferred geometries; the orbital-overlap mechanism will be illustrated with explicit charge-density difference plots. revision: yes
Circularity Check
No circularity: DMC benchmark is independent validation for functional selection
full rationale
The derivation chain uses DMC calculations as an external, first-principles benchmark to identify which DFT functional (SCAN+rVV10) best matches equilibrium separations and energetics. The selected functional is then applied to compute the thickness-dependent stacking sequence and band-gap trends. This is a standard cross-method validation workflow with no reduction of outputs to inputs by construction, no self-citation load-bearing steps, and no fitted parameters renamed as predictions. The abstract and described methodology treat DMC results as independent reference data rather than tautological inputs.
Axiom & Free-Parameter Ledger
Reference graph
Works this paper leans on
-
[1]
& Tom´ anek, D
Zhu, Z., Guan, J. & Tom´ anek, D. Strain-induced metal-semiconductor transition in monolay- ers and bilayers of gray arsenic: A computational study. Phys. Rev. B91, 161404 (2015)
2015
-
[2]
& Ciraci, S
Kecik, D., Durgun, E. & Ciraci, S. Stability of single-layer and multilayer arsenene and their mechanical and electronic properties. Phys. Rev. B94, 205409 (2016)
2016
-
[3]
Ersan, F. et al. Two-dimensional pnictogens: A review of recent progresses and future research directions. Appl. Phys. Rev.6, 021308 (2019)
2019
-
[4]
& Ezawa, M
Kamal, C. & Ezawa, M. Arsenene: Two-dimensional buckled and puckered honeycomb arsenic systems. Phys. Rev. B91, 085423 (2015)
2015
-
[5]
Shah, J., Wang, W., Sohail, H. M. & Uhrberg, R. Experimental evidence of monolayer arsenene: an exotic 2D semiconducting material. 2D Mater.7, 025013 (2020)
2020
-
[6]
Density of states calculation for crystalline As and Sb
Bullett, D. Density of states calculation for crystalline As and Sb. Solid State Commun.17, 965–967 (1975)
1975
-
[7]
& Vigneron, J.-P
Gonze, X., Michenaud, J.-P. & Vigneron, J.-P. First-principles study of As, Sb, and Bi electronic properties. Phys. Rev. B41, 11827 (1990)
1990
-
[8]
Xu, J., Wang, E., Ting, C. & Su, W. Tight-binding theory of the electronic structures for rhombohedral semimetals. Phys. Rev. B48, 17271 (1993)
1993
-
[9]
Zhao, L. et al. Magnetotransport properties in a compensated semimetal gray arsenic. Phys. Rev. B95, 115119 (2017)
2017
-
[10]
E., Merino, G
Arcudia, J., Kempt, R., Cifuentes-Quintal, M. E., Merino, G. & Heine, T. Blue phosphorene bilayer is a two-dimensional metal–and an unambiguous classification scheme for buckled hexagonal bilayers. Phys. Rev. Lett.125, 196401 (2020)
2020
-
[11]
Kadioglu, Y. et al. Diffusion quantum monte carlo and density functional calculations of the structural stability of bilayer arsenene. J. Chem. Phys.148, 214706 (2018). 16
2018
-
[12]
& Barrett, C
Schiferl, D. & Barrett, C. The crystal structure of arsenic at 4.2, 78 and 299 K. J. Appl. Crystallogr.2, 30–36 (1969)
1969
-
[13]
Kim, J. et al. QMCPACK: an open source ab initio quantum Monte Carlo package for the electronic structure of atoms, molecules and solids. J. Phys.: Condens. Matter30, 195901 (2018)
2018
-
[14]
Kent, P. R. C. et al. QMCPACK: Advances in the development, efficiency, and application of auxiliary field and real-space variational and diffusion quantum Monte Carlo. J. Chem. Phys. 152, 174105 (2020)
2020
-
[15]
Giannozzi, P. et al. QUANTUM ESPRESSO: a modular and open-source software project for quantum simulations of materials. J. Phys.: Condens. Matter21, 395502 (2009)
2009
-
[16]
Monkhorst, H. J. & Pack, J. D. Special points for Brillouin-zone integrations. Phys. Rev. B 13, 5188 (1976)
1976
-
[17]
Wang, G. et al. A new generation of effective core potentials from correlated calculations: 4s and 4p main group elements and first row additions. J. Chem. Phys.151, 144110 (2019)
2019
-
[18]
Lin, C., Zong, F. H. & Ceperley, D. M. Twist-averaged boundary conditions in continuum quantum Monte Carlo algorithms. Phys. Rev. E64, 016702 (2001)
2001
-
[19]
J., Toulouse, J., Filippi, C., Sorella, S
Umrigar, C. J., Toulouse, J., Filippi, C., Sorella, S. & Hennig, R. G. Alleviation of the fermion-sign problem by optimization of many-body wave functions. Phys. Rev. Lett.98, 110201 (2007)
2007
-
[20]
M., Mitas, L., Needs, R
Foulkes, W. M., Mitas, L., Needs, R. & Rajagopal, G. Quantum monte carlo simulations of solids. Rev. Mod. Phys.73, 33 (2001)
2001
-
[21]
& Hafner, J
Kresse, G. & Hafner, J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B47, 558–561 (1993)
1993
-
[22]
& Furthm¨ uller, J
Kresse, G. & Furthm¨ uller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B54, 11169–11186 (1996)
1996
-
[23]
Bl¨ ochl, P. E. Projector augmented-wave method. Phys. Rev. B50, 17953–17979 (1994)
1994
-
[24]
& Joubert, D
Kresse, G. & Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B59, 1758–1775 (1999)
1999
-
[25]
Perdew, J. P. & Zunger, A. Self-interaction correction to density-functional approximations for many-electron systems. Phys. Rev. B23, 5048 (1981). 17
1981
-
[26]
Semiempirical GGA-type density functional constructed with a long-range dis- persion correction
Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dis- persion correction. J. Comput. Chem.27, 1787–1799 (2006)
2006
-
[27]
Klimeˇ s, J., Bowler, D. R. & Michaelides, A. Chemical accuracy for the van der Waals density functional. J. Phys.: Condens. Matter22, 022201 (2009)
2009
-
[28]
Peng, H., Yang, Z.-H., Perdew, J. P. & Sun, J. Versatile van der Waals density functional based on a meta-generalized gradient approximation. Phys. Rev. X6, 041005 (2016)
2016
-
[29]
Blaiszik, B. et al. The materials data facility: Data services to advance materials science research. JOM68, 2045–2052 (2016). URLhttps://doi.org/10.1007/s11837-016-2001-3
-
[30]
Blaiszik, B. et al. A data ecosystem to support machine learning in materials science. MRS Commun.9, 1125–1133 (2019). URLhttps://doi.org/10.1557/mrc.2019.118. 18
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.