SALMON 2.3: Implementation of divide-and-conquer ground-state initialization for large-scale real-time TDDFT
Pith reviewed 2026-06-26 14:01 UTC · model grok-4.3
The pith
Divide-and-conquer DFT prepares ground states for real-time TDDFT in linear time with system size.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
The DC-DFT self-consistent-field procedure exhibits linear scaling with system size, addressing a major bottleneck in large-scale electron-dynamics simulations while retaining the robustness and broad applicability of SALMON's established time-propagation scheme. Reconstructed global orbitals from the divide-and-conquer subsystems serve directly as initial states for the standard real-time, real-space TDDFT module.
What carries the argument
Divide-and-conquer DFT (DC-DFT) scheme with postprocessing reconstruction of spatially extended Kohn-Sham orbitals, which supplies the initial wave functions for the conventional real-time TDDFT propagation.
If this is right
- Ground-state preparation time grows linearly rather than cubically, so total wall time for real-time TDDFT becomes dominated by the propagation step even for systems with thousands of atoms.
- The same workflow applies unchanged to disordered materials, liquids, nanostructures, and heterogeneous systems because the DC-DFT step and reconstruction do not rely on periodicity.
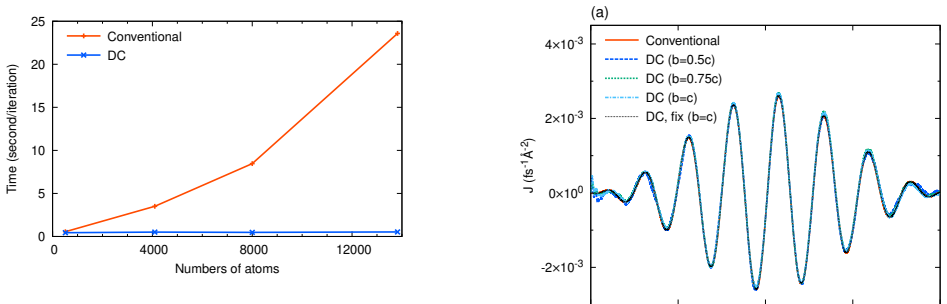
- Parallel scaling tests on Fugaku confirm that the DC-DFT phase itself scales weakly with the number of MPI processes, preserving the code's existing massive-parallelism strategy.
- Input and output formats remain compatible with the standard SALMON time-propagation module, so existing simulation scripts require only a change in the ground-state initialization flag.
Where Pith is reading between the lines
- If the reconstruction step can be made fully automatic and black-box, the method could be adopted by other real-space TDDFT codes without requiring users to tune subsystem partitioning.
- The linear scaling opens the possibility of running real-time TDDFT on entire device-scale models rather than periodic supercells, provided memory per node remains manageable.
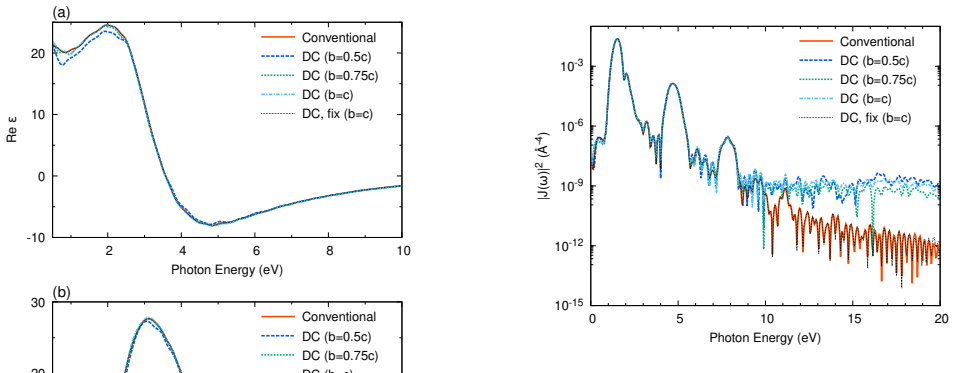
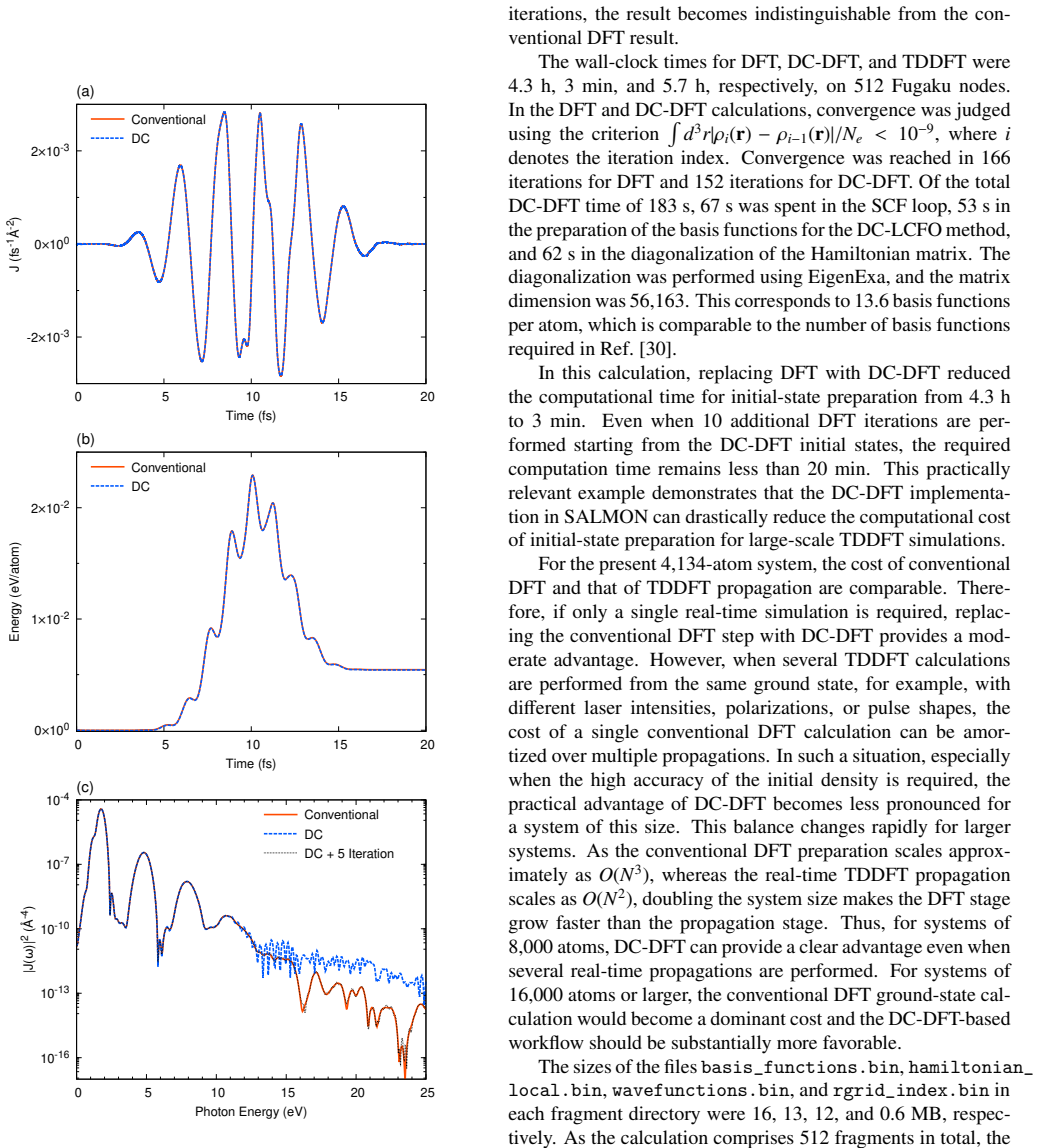
- Accuracy tests on 4134-atom liquid water already show practical usability; extending the same test to a heterogeneous interface would immediately reveal whether interface states are faithfully captured.
Load-bearing premise
The reconstructed global orbitals from the subsystem calculations remain accurate enough that errors do not propagate significantly during the subsequent real-time TDDFT evolution.
What would settle it
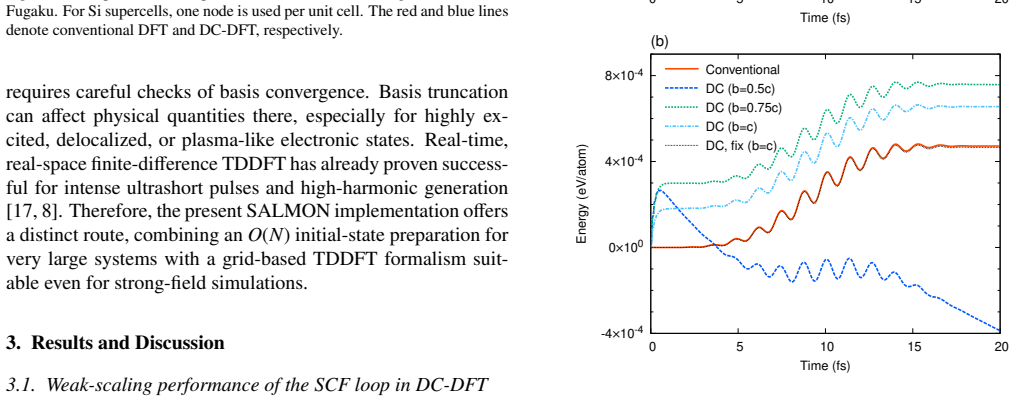
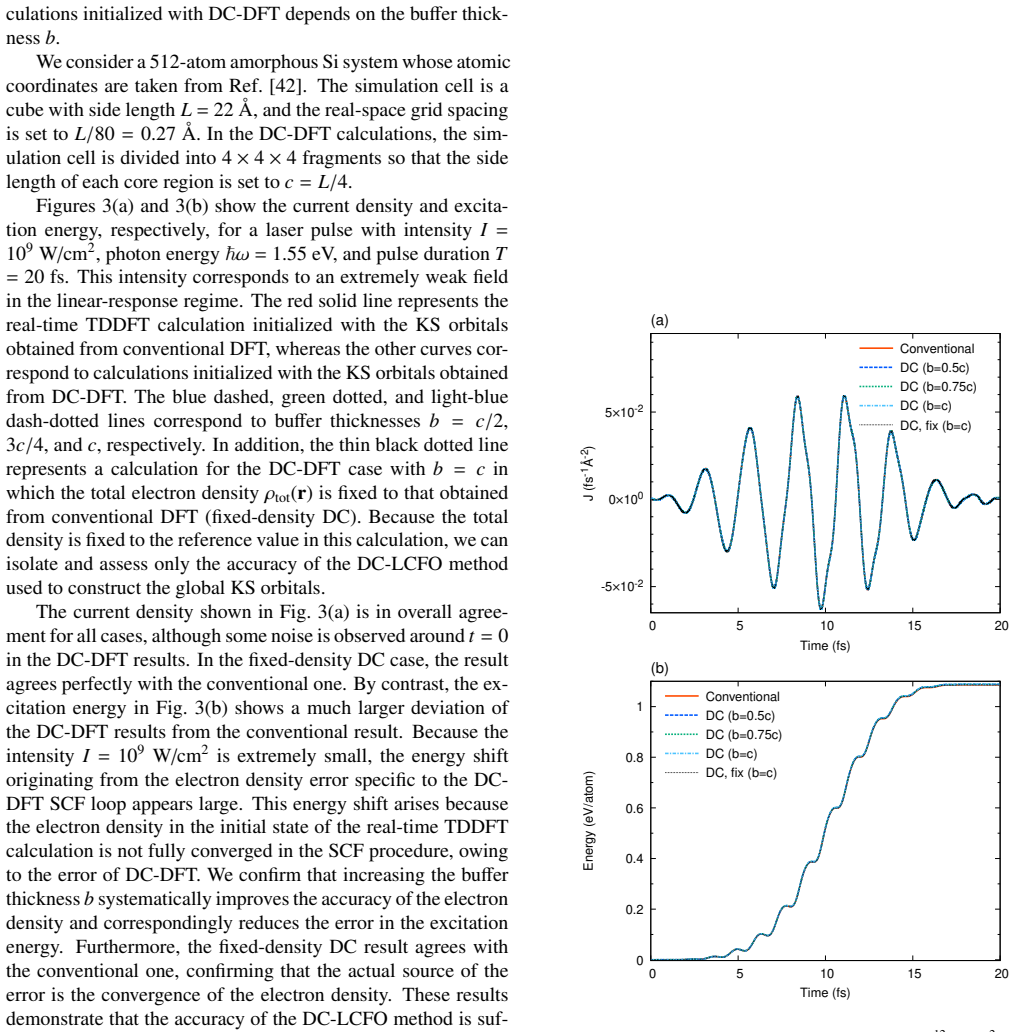
Run the new workflow on a 512-atom amorphous silicon system and compare the time-dependent dipole response or absorption spectrum against a reference calculation started from a conventional ground-state solver; any large discrepancy would falsify the claim that the reconstruction introduces no significant errors.
Figures

read the original abstract
In large-scale real-time time-dependent density functional theory (TDDFT), preparing the ground-state electronic structure can be more expensive than the subsequent time propagation, limiting simulations of nonequilibrium electron dynamics in realistic systems containing thousands of atoms. This bottleneck is especially important for disordered materials, liquids, nanostructures, and heterogeneous condensed-matter systems, where nonlinear and strong-field phenomena such as high-harmonic generation and light-induced phase transitions require explicit real-time treatment. SALMON is an open-source first-principles code for light-matter interaction simulations based on real-time TDDFT on real-space grids, supporting massively parallel calculations with MPI combined with OpenMP or GPU acceleration. In SALMON 2.3, we implement a divide-and-conquer density functional theory (DC-DFT) scheme and combine it with a postprocessing method that reconstructs spatially extended Kohn-Sham orbitals of the entire system. These reconstructed global orbitals are used directly as initial states for the standard real-time, real-space TDDFT module of SALMON. The resulting workflow connects efficient DC-DFT ground-state preparation to conventional real-time TDDFT. The DC-DFT self-consistent-field procedure exhibits linear scaling with system size, addressing a major bottleneck in large-scale electron-dynamics simulations while retaining the robustness and broad applicability of SALMON's established time-propagation scheme. We describe the computational procedure, parallelization strategy, and input/output design. Weak-scaling tests using Si supercells on Fugaku confirm the linear-scaling behavior. Accuracy tests for a 512-atom amorphous Si system and a bulk H2O liquid system containing 4134 atoms demonstrate that the workflow enables practical large-scale real-time TDDFT simulations.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript describes the implementation of a divide-and-conquer DFT (DC-DFT) scheme in SALMON 2.3 for ground-state preparation in large-scale real-time TDDFT. It combines DC-DFT SCF with a postprocessing reconstruction of global Kohn-Sham orbitals from subsystems; these orbitals initialize the standard real-space real-time TDDFT propagation module. Weak-scaling tests on Fugaku using Si supercells demonstrate linear scaling with system size. Accuracy tests on a 512-atom amorphous Si system and a 4134-atom bulk H2O liquid system are presented to show that the workflow enables practical simulations of nonequilibrium dynamics in large systems.
Significance. If the reconstruction step produces initial states whose errors remain negligible for the subsequent nonlinear dynamics, energy conservation, and spectra, the work would meaningfully extend real-time TDDFT applicability to systems with thousands of atoms by removing the dominant ground-state cost while retaining SALMON's established propagation robustness. The linear-scaling demonstration on Fugaku and the open-source code release constitute concrete strengths.
major comments (2)
- [Accuracy tests (abstract and results section)] Accuracy tests (abstract and results section): no quantitative metrics are supplied for reconstruction fidelity on the 512-atom a-Si or 4134-atom H2O systems (e.g., orbital overlap deviations, ||ρ_recon − ρ_conv||, initial total-energy difference, or propagation drift relative to conventional SCF initialization). Because the central claim is that the reconstructed orbitals serve as sufficiently accurate initial conditions without compromising the TDDFT scheme, the absence of these numbers leaves the load-bearing assumption unverified.
- [Weak-scaling tests on Fugaku] Weak-scaling tests on Fugaku: the reported linear scaling of the DC-DFT SCF is useful, yet the manuscript does not quantify the additional wall-time or parallel overhead introduced by the postprocessing reconstruction step itself; without this breakdown it is difficult to confirm that the full workflow (SCF + reconstruction) remains linearly scaling and removes the stated bottleneck.
minor comments (1)
- [Computational procedure and parallelization] The description of the parallelization strategy and I/O design for the reconstruction step would benefit from a short pseudocode or data-flow diagram to clarify how subsystem orbitals are assembled into global orbitals on distributed memory.
Simulated Author's Rebuttal
We thank the referee for the constructive comments on our manuscript. We respond to each major comment below and indicate the changes we will make in revision.
read point-by-point responses
-
Referee: [Accuracy tests (abstract and results section)] Accuracy tests (abstract and results section): no quantitative metrics are supplied for reconstruction fidelity on the 512-atom a-Si or 4134-atom H2O systems (e.g., orbital overlap deviations, ||ρ_recon − ρ_conv||, initial total-energy difference, or propagation drift relative to conventional SCF initialization). Because the central claim is that the reconstructed orbitals serve as sufficiently accurate initial conditions without compromising the TDDFT scheme, the absence of these numbers leaves the load-bearing assumption unverified.
Authors: We agree that quantitative metrics are required to substantiate the claim that reconstruction errors remain negligible for the subsequent TDDFT dynamics. In the revised manuscript we will add explicit numerical values (orbital overlaps, ||ρ_recon − ρ_conv||, initial total-energy differences, and observed propagation drift relative to conventional SCF) for both the 512-atom a-Si and 4134-atom H2O test cases. revision: yes
-
Referee: [Weak-scaling tests on Fugaku] Weak-scaling tests on Fugaku: the reported linear scaling of the DC-DFT SCF is useful, yet the manuscript does not quantify the additional wall-time or parallel overhead introduced by the postprocessing reconstruction step itself; without this breakdown it is difficult to confirm that the full workflow (SCF + reconstruction) remains linearly scaling and removes the stated bottleneck.
Authors: We acknowledge that the timing data presented focused on the DC-DFT SCF phase and did not isolate the reconstruction overhead. In the revised manuscript we will include a separate timing breakdown of the reconstruction postprocessing step, together with its contribution to total wall time and parallel efficiency, to confirm that the end-to-end workflow retains linear scaling. revision: yes
Circularity Check
No circularity: standard DC-DFT implementation with external validation
full rationale
The paper presents an engineering implementation of established divide-and-conquer DFT for ground-state preparation, followed by a postprocessing reconstruction step whose outputs feed into an existing real-time TDDFT propagator. Linear scaling is asserted as a property of DC-DFT and confirmed by weak-scaling benchmarks on Si supercells rather than derived from the result itself. Accuracy is demonstrated via explicit tests on 512-atom a-Si and 4134-atom H2O systems, with no fitted parameters, self-referential definitions, or load-bearing self-citations that reduce the central workflow to its own inputs. The reconstruction step is described procedurally without any equation or claim that equates the output to the input by construction.
Axiom & Free-Parameter Ledger
axioms (1)
- domain assumption Standard Kohn-Sham DFT on subsystems can be combined via divide-and-conquer to approximate the full-system ground state
Reference graph
Works this paper leans on
-
[1]
P. Hohenberg, W. Kohn, Inhomogeneous elec- tron gas, Physical Review 136 (1964) B864–B871. doi:10.1103/PhysRev.136.B864
-
[2]
W. Kohn, L. J. Sham, Self-consistent equations includ- ing exchange and correlation effects, Physical Review 140 (1965) A1133–A1138. doi:10.1103/PhysRev.140.A1133
-
[3]
E. Runge, E. K. U. Gross, Density-functional theory for time-dependent systems, Physical Review Letters 52 (1984) 997–1000. doi:10.1103/PhysRevLett.52.997. URLhttps://journals.aps.org/prl/pdf/10. 1103/PhysRevLett.52.997https://link.aps. org/doi/10.1103/PhysRevLett.52.997
-
[4]
Burke, Perspective on density functional theory, The Journal of Chemical Physics 136 (4 2012)
K. Burke, Perspective on density functional theory, The Journal of Chemical Physics 136 (4 2012). doi:10.1063/1.4704546
-
[5]
R. Jones, Density functional theory: Its origins, rise to prominence, and future, Reviews of Modern Physics 87 (2015) 897–923. doi:10.1103/RevModPhys.87.897
-
[6]
C. A. Ullrich, Time-Dependent Density-Functional The- ory: Concepts and Applications, Oxford University Press, Oxford, 2011
2011
-
[7]
M. A. L. Marques, C. A. Ullrich, F. Nogueira, A. Rubio, K. Burke, E. K. U. Gross (Eds.), Time-Dependent Density Functional Theory, V ol. 706 of Lecture Notes in Physics, Springer, Berlin and Heidelberg, 2006
2006
-
[8]
S. A. Sato, H. Hübener, U. D. Giovannini, A. Ru- bio, Technical review: Time-dependent density functional theory for attosecond physics ranging from gas-phase to solids, npj Computational Materials 11 (2025) 233. doi:10.1038/s41524-025-01715-1
-
[9]
J. VandeV ondele, U. Borštnik, J. Hutter, Linear scal- ing self-consistent field calculations with millions of atoms in the condensed phase, Journal of Chemi- cal Theory and Computation 8 (2012) 3565–3573. doi:10.1021/ct200897x
-
[10]
A. Nakata, J. S. Baker, S. Y . Mujahed, J. T. L. Poul- ton, S. Arapan, J. Lin, Z. Raza, S. Yadav, L. Truflandier, T. Miyazaki, D. R. Bowler, Large scale and linear scal- ing dft with the conquest code, The Journal of Chemical Physics 152 (4 2020). doi:10.1063/5.0005074
-
[11]
J. C. A. Prentice, J. Aarons, J. C. Womack, A. E. A. Allen, L. Andrinopoulos, L. Anton, R. A. Bell, A. Bhan- dari, G. A. Bramley, R. J. Charlton, R. J. Clements, D. J. Cole, G. Constantinescu, F. Corsetti, S. M.-M. Dubois, K. K. B. Duff, J. M. Escartín, A. Greco, Q. Hill, L. P. Lee, E. Linscott, D. D. O’Regan, M. J. S. Phipps, L. E. Ratcliff, Álvaro Ruiz ...
-
[12]
T. J. Zuehlsdorff, N. D. M. Hine, J. S. Spencer, N. M. Harrison, D. J. Riley, P. D. Haynes, Linear-scaling time- dependent density-functional theory in the linear response formalism, The Journal of Chemical Physics 139 (8 2013). doi:10.1063/1.4817330
-
[13]
C. O’Rourke, D. R. Bowler, Linear scaling density ma- trix real time tddft: Propagator unitarity and matrix trun- cation, The Journal of Chemical Physics 143 (9 2015). doi:10.1063/1.4919128
-
[14]
A. Bussy, J. Hutter, Efficient and low-scaling linear- response time-dependent density functional theory imple- mentation for core-level spectroscopy of large and peri- odic systems, Physical Chemistry Chemical Physics 23 (2021) 4736–4746. doi:10.1039/D0CP06164F
-
[15]
Y . Takimoto, F. D. Vila, J. J. Rehr, Real-time time-dependent density functional theory approach for frequency-dependent nonlinear optical response in pho- tonic molecules, The Journal of Chemical Physics 127 (10 2007). doi:10.1063/1.2790014
-
[16]
J. Feng, J. Chen, X. Zhang, J. Liu, X. Qin, L. Wan, S. Chen, W. Wu, B. Hou, Y . Lin, Y . Zhang, Z. Zhang, Y . Hu, W. Jia, H. An, J. Yang, W. Hu, Million-atom ab ini- tio electron dynamics: Discontinuous galerkin real-time time-dependent density functional theory, in: Proceedings of the International Conference for High Performance Computing, Networking, S...
-
[17]
M. Noda, S. A. Sato, Y . Hirokawa, M. Uemoto, T. Takeuchi, S. Yamada, A. Yamada, Y . Shinohara, M. Yamaguchi, K. Iida, I. Floss, T. Otobe, K.-M. K.-M. Lee, K. Ishimura, T. Boku, G. F. Bertsch, K. Nobusada, K. Yabana, Salmon: Scalable ab-initio light–matter simulator for optics and nanoscience, Com- puter Physics Communications 235 (2019) 356–365. doi:10.1...
-
[18]
Y . Hirokawa, A. Yamada, S. Yamada, M. Noda, M. Uemoto, T. Boku, K. Yabana, Large-scale ab initio simulation of light–matter interaction at the atomic scale in fugaku, The International Journal of High Perfor- mance Computing Applications 36 (2022) 182–197. doi:10.1177/10943420211065723. URLhttp://journals.sagepub.com/doi/10. 1177/10943420211065723
-
[19]
S. Goedecker, Linear scaling electronic structure meth- ods, Reviews of Modern Physics 71 (1999) 1085–1123. doi:10.1103/RevModPhys.71.1085
-
[20]
D. R. Bowler, T. Miyazaki, {{O}(N)} methods in electronic structure calculations, Reports on Progress in Physics 75 (2012) 036503. doi:10.1088/0034- 4885/75/3/036503. URLhttps://iopscience.iop.org/article/10. 1088/0034-4885/75/3/036503
-
[21]
W. Yang, Direct calculation of electron density in density- functional theory, Physical Review Letters 66 (1991) 1438–1441. doi:10.1103/PhysRevLett.66.1438
-
[22]
W. Yang, T.-S. Lee, A density-matrix divide-and-conquer approach for electronic structure calculations of large molecules, The Journal of Chemical Physics 103 (1995) 5674–5678. doi:10.1063/1.470549
-
[23]
F. Shimojo, R. K. Kalia, A. Nakano, P. Vashishta, Embedded divide-and-conquer algorithm on hierarchical real-space grids: Parallel molecular dynamics simula- tion based on linear-scaling density functional theory, Computer Physics Communications 167 (2005) 151–164. doi:10.1016/j.cpc.2005.01.005
-
[24]
F. Shimojo, R. K. Kalia, A. Nakano, P. Vashishta, Divide-and-conquer density functional theory on hi- erarchical real-space grids: Parallel implementation and applications, Physical Review B 77 (2008) 1–12. doi:10.1103/PhysRevB.77.085103
-
[25]
N. Ohba, S. Ogata, T. Kouno, T. Tamura, R. Kobayashi, Linear scaling algorithm of real-space density functional theory of electrons with correlated overlapping domains, Computer Physics Communications 183 (2012) 1664–
2012
-
[26]
URLhttp://dx.doi.org/10.1016/j.cpc.2012
doi:10.1016/j.cpc.2012.03.004. URLhttp://dx.doi.org/10.1016/j.cpc.2012. 03.004
-
[27]
F. Shimojo, S. Hattori, R. K. Kalia, M. Kunaseth, W. Mou, A. Nakano, K. I. Nomura, S. Ohmura, P. Rajak, K. Shima- mura, P. Vashishta, A divide-conquer-recombine algorith- mic paradigm for large spatiotemporal quantum molecular dynamics simulations, Journal of Chemical Physics 140 (2014). doi:10.1063/1.4869342
-
[28]
W. Kohn, Density functional and density matrix method scaling linearly with the number of atoms, Physical Review Letters 76 (1996) 3168–3171. doi:10.1103/PhysRevLett.76.3168
-
[29]
E. Prodan, W. Kohn, Nearsightedness of electronic matter, Proceedings of the National Academy of Sciences 102 (2005) 11635–11638. doi:10.1073/pnas.0505436102. URLhttp://www.pnas.org/cgi/doi/10.1073/ pnas.0505436102 12
-
[30]
S. Yamada, F. Shimojo, R. Akashi, S. Tsuneyuki, Efficient method for calculating spatially extended electronic states of large systems with a divide-and- conquer approach, Physical Review B 95 (2017) 045106. doi:10.1103/PhysRevB.95.045106. URLhttps://link.aps.org/doi/10.1103/ PhysRevB.95.045106http://link.aps.org/doi/ 10.1103/PhysRevB.95.045106
-
[31]
N. Troullier, J. L. Martins, Efficient pseudopotentials for plane-wave calculations, Physical Review B 43 (1991) 1993–2006. doi:10.1103/PhysRevB.43.1993. URLhttps://journals.aps.org/prb/pdf/10. 1103/PhysRevB.43.1993https://link.aps.org/ doi/10.1103/PhysRevB.43.1993
-
[32]
J. P. Perdew, A. Zunger, Self-interaction correction to density-functional approximations for many-electron systems, Physical Review B 23 (1981) 5048–5079. doi:10.1103/PhysRevB.23.5048. URLhttps://link.aps.org/doi/10.1103/ PhysRevB.23.5048
-
[33]
T. Fukaya, T. Imamura, Performance evaluation of the eigen exa eigensolver on oakleaf-fx: Tridi- agonalization versus pentadiagonalization, in: 2015 IEEE International Parallel and Distributed Process- ing Symposium Workshop, IEEE, 2015, pp. 960–969. doi:10.1109/IPDPSW.2015.128. [34]https://www.r-ccs.riken.jp/labs/lpnctrt/ projects/eigenexa, EigenExa website
-
[34]
T. Sakurai, H. Sugiura, A projection method for gener- alized eigenvalue problems using numerical integration, Journal of Computational and Applied Mathematics 159 (2003) 119–128. doi:10.1016/S0377-0427(03)00565-X
-
[35]
X. Andrade, J. Alberdi-Rodriguez, D. A. Strubbe, M. J. T. Oliveira, F. Nogueira, A. Castro, J. Muguerza, A. Ar- ruabarrena, S. G. Louie, A. Aspuru-Guzik, A. Rubio, M. A. L. Marques, Time-dependent density-functional theory in massively parallel computer architectures: the octopus project, Journal of Physics: Condensed Matter 24 (2012) 233202. doi:10.1088/...
-
[36]
K. Hanasaki, S. Luber, Development of real-time tddft program with k -point sampling and dft+u in a gaus- sian and plane waves framework, Journal of Chem- ical Theory and Computation 21 (2025) 1879–1891. doi:10.1021/acs.jctc.4c01515
-
[37]
Lopata, N
K. Lopata, N. Govind, Modeling fast electron dynamics with real-time time-dependent density functional theory: Application to small molecules and chromophores, Jour- nal of Chemical Theory and Computation 7 (2011) 1344–
2011
-
[38]
doi:10.1021/ct200137z
-
[39]
L. Lin, J. Lu, L. Ying, W. E, Adaptive local basis set for kohn–sham density functional theory in a discon- tinuous galerkin framework i: Total energy calculation, Journal of Computational Physics 231 (2012) 2140–2154. doi:10.1016/j.jcp.2011.11.032
-
[40]
G. Zhang, L. Lin, W. Hu, C. Yang, J. E. Pask, Adap- tive local basis set for kohn–sham density functional the- ory in a discontinuous galerkin framework ii: Force, vibration, and molecular dynamics calculations, Jour- nal of Computational Physics 335 (2017) 426–443. doi:10.1016/j.jcp.2016.12.052
-
[41]
V . L. Deringer, N. Bernstein, A. P. Bartók, M. J. Cliffe, R. N. Kerber, L. E. Marbella, C. P. Grey, S. R. Elliott, G. Csányi, Realistic atomistic structure of amorphous sil- icon from machine-learning-driven molecular dynamics, The Journal of Physical Chemistry Letters 9 (2018) 2879–
2018
-
[42]
doi:10.1021/acs.jpclett.8b00902
-
[43]
M. J. Abraham, T. Murtola, R. Schulz, S. Páll, J. C. Smith, B. Hess, E. Lindahl, Gromacs: High performance molec- ular simulations through multi-level parallelism from lap- tops to supercomputers, SoftwareX 1-2 (2015) 19–25. doi:10.1016/j.softx.2015.06.001. [44]https://www.gromacs.org, GROMACS website. 13
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.