Computationally guided modifications of CviUPO to improve catalytic activity
Pith reviewed 2026-06-26 12:48 UTC · model grok-4.3
The pith
Replacing the heme-anchoring cysteine with histidine in CviUPO lowers the energy barriers for catalysis in QM/MM simulations.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
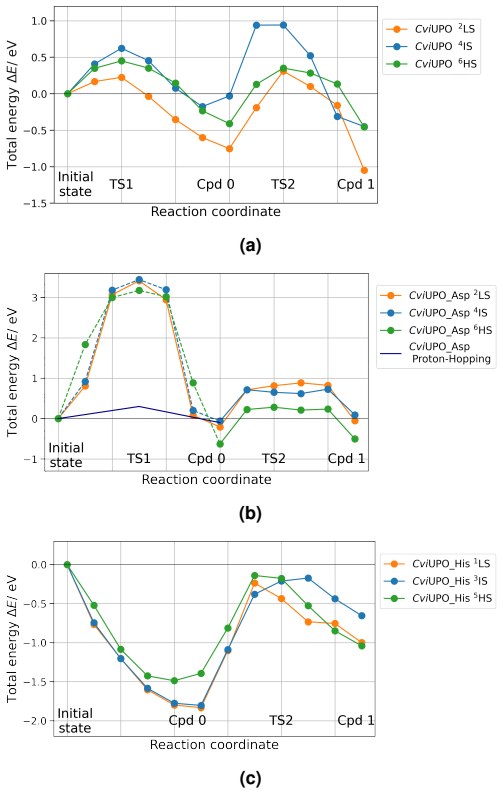
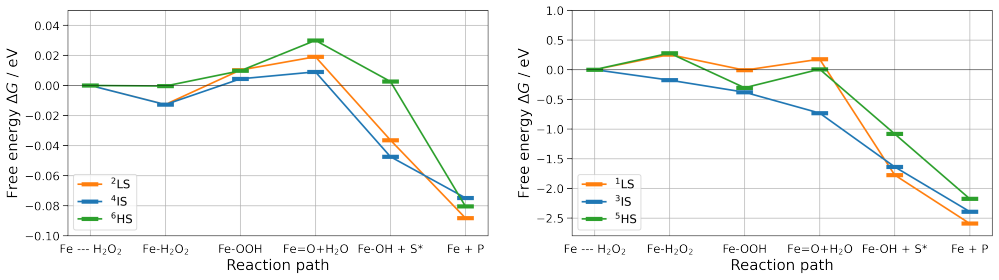
QM/MM NEB simulations show that the cysteine-to-histidine mutation at the heme anchor decreases the energy barriers significantly while the glutamic-to-aspartic acid mutation increases them; the histidine change may also shift the enzyme toward peroxidase behavior, an outcome the calculations cannot rule out.
What carries the argument
Quantum Mechanics/Molecular Mechanics Nudged Elastic Band (NEB) simulations that compute changes in activation energy barriers after single-residue mutations close to the active center.
If this is right
- The cysteine-to-histidine mutation is predicted to improve catalytic performance provided the enzyme retains peroxygenase reactivity.
- The glutamic-to-aspartic acid mutation is predicted to impair activity by raising the reaction barrier.
- Spin states and the degree of active-pocket hydration are identified as factors that control barrier height in the catalytic cycle.
- Efficient engineering of UPOs requires iterative combination of simulation predictions with experimental tests of substrate specificity and stability.
Where Pith is reading between the lines
- The same computational protocol could be applied to other UPO variants to identify stabilizing mutations at the heme anchor without exhaustive library screening.
- If the histidine mutant retains peroxygenase function, it would expand the range of conditions under which these enzymes can operate before inactivation occurs.
- Testing whether the mutation alters the preferred spin state during catalysis would directly address one of the paper's highlighted mechanistic factors.
- The results point to heme-ligating residues as a general hotspot worth prioritizing in future rounds of UPO redesign.
Load-bearing premise
The simulations correctly describe the catalytic mechanism and the histidine mutation leaves the enzyme's peroxygenase character intact rather than converting it to a peroxidase.
What would settle it
Direct measurement of catalytic turnover rate and product profile for the histidine-mutant CviUPO acting on a saturated hydrocarbon substrate, compared with the wild-type enzyme under identical peroxide conditions.
Figures

read the original abstract
Unspecific peroxygenases (UPOs) are promising biocatalysts that selectively oxyfunctionalize saturated hydrocarbons using only hydrogen peroxide as a co-substrate. Peroxide-induced enzyme inactivation makes targeted enzyme engineering essential to mitigate this effect and also enhance catalytic performance. To meet this need, systematic approaches are used, including extensive database studies for rational enzyme design, as well as computational enzyme engineering. In this study, we followed the latter strategy and explored the possibility for computationally-guided modification of UPOs. Specifically, our focus was on uncovering the influence of active site amino acids on the catalytic activity of the enzyme CviUPO. Two mutations were introduced close to the active center, and the changes in the energy barriers leading to the activated complex were investigated in detail by Quantum Mechanics/Molecular Mechanics Nudged Elastic Band simulations. Our studies revealed that a change of the glutamic acid, assisting the catalytic cycle, by the shorter aspartic acid, leads to an increased reaction barrier, probably decreasing the catalytic activity of the enzyme. Exchanging the heme-anchoring cysteine group by a histidine exhibited promising behavior as the energy barriers decreased significantly. However, it is possible that the histidine modification also alters the reaction behavior of the peroxygenase, turning it into a peroxidase, an aspect that so far could not be confirmed beyond doubt. Simulations alone cannot conclusively determine whether substrate specificity and reactivity are maintained in the modifications tested. Nevertheless, our results highlight the importance of spin states and active pocket hydration for the catalytic reaction and demonstrate why a synergistic approach of theoretical predictions and experimental verifications is required for efficient enzyme engineering.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript uses QM/MM NEB simulations to examine the effects of two active-site mutations in CviUPO on the barriers to the activated complex. Replacement of the assisting glutamic acid by aspartic acid is reported to raise the barrier, while replacement of the heme-ligating cysteine by histidine is reported to lower the barriers substantially; the authors note that the histidine change may convert the enzyme to peroxidase behavior but state that this could not be confirmed.
Significance. If the peroxygenase mechanism is preserved and the barrier changes are robust, the work would illustrate how proximal residues and spin-state/hydration effects modulate UPO catalysis and would support the value of QM/MM NEB for guiding peroxygenase engineering. The explicit caveat about possible mechanism switching and the call for experimental verification are appropriate.
major comments (2)
- [Abstract and histidine-mutation results] Abstract and the histidine-mutation results section: the NEB paths for the Cys-to-His mutant follow only the original peroxygenase cycle; no barriers or reaction coordinates are supplied for the competing O–O cleavage or Compound-I formation channels that would be required to rule out a switch to peroxidase reactivity, even though the abstract itself flags this possibility.
- [Results and Methods (NEB)] Results and Methods sections on the NEB calculations: the manuscript states directional changes in barriers (“increased,” “decreased significantly”) but supplies neither the numerical barrier heights, their standard errors, nor convergence or spin-state sampling details, so the magnitude and statistical reliability of the reported effects cannot be evaluated.
minor comments (1)
- [Abstract] The abstract would benefit from a single sentence stating the computed barrier changes (in kcal/mol) if those values appear in the main text or SI.
Simulated Author's Rebuttal
We thank the referee for their constructive comments. We address each major point below, indicating planned revisions where appropriate.
read point-by-point responses
-
Referee: [Abstract and histidine-mutation results] Abstract and the histidine-mutation results section: the NEB paths for the Cys-to-His mutant follow only the original peroxygenase cycle; no barriers or reaction coordinates are supplied for the competing O–O cleavage or Compound-I formation channels that would be required to rule out a switch to peroxidase reactivity, even though the abstract itself flags this possibility.
Authors: We acknowledge that the NEB calculations addressed only the peroxygenase reaction coordinate. The abstract already states that a switch to peroxidase behavior could not be confirmed. We will revise the abstract and results to state explicitly that the reported barrier lowering applies solely to the peroxygenase pathway and that alternative channels were not computed. This limitation follows from the study scope; the computational evidence still indicates a lowered barrier along the original cycle, supporting the call for experimental verification. revision: partial
-
Referee: [Results and Methods (NEB)] Results and Methods sections on the NEB calculations: the manuscript states directional changes in barriers (“increased,” “decreased significantly”) but supplies neither the numerical barrier heights, their standard errors, nor convergence or spin-state sampling details, so the magnitude and statistical reliability of the reported effects cannot be evaluated.
Authors: We agree that numerical barrier values and methodological details would improve evaluability. In the revised manuscript we will report the specific NEB barrier heights, spin-state sampling protocol, and convergence criteria employed in the QM/MM calculations. revision: yes
- Providing barriers or reaction coordinates for the competing O–O cleavage and Compound-I formation channels in the Cys-to-His mutant, which would require new QM/MM NEB simulations beyond the original study.
Circularity Check
No circularity: results from independent QM/MM simulations
full rationale
The paper reports direct QM/MM NEB calculations of energy barriers for CviUPO wild-type and two point mutants (Glu-to-Asp, Cys-to-His). No parameters are fitted to experimental outcomes, no self-citations underpin the central barrier comparisons, and the derivation chain consists of standard electronic-structure plus classical MD steps whose outputs are not definitionally equivalent to their inputs. The abstract itself flags the open question of peroxygenase vs. peroxidase behavior, confirming the result is not smuggled in by assumption.
Axiom & Free-Parameter Ledger
axioms (1)
- domain assumption QM/MM NEB simulations with the chosen level of theory and spin states correctly identify the lowest-energy reaction paths and barrier heights for the catalytic cycle.
Reference graph
Works this paper leans on
-
[1]
Wu, S., Snajdrova, R., Moore, J. C., Baldenius, K. & Bornscheuer, U. T. Biocatalysis: Enzymatic synthesis for industrial applications.Angewandte Chemie Int. Ed.60, 88–119, DOI: 10.1002/ANIE.202006648 (2021)
-
[2]
Hofrichter, M. & Ullrich, R. Oxidations catalyzed by fungal peroxygenases.Curr. Opin. Chem. Biol.19, 116–125, DOI: 10.1016/j.cbpa.2014.01.015 (2014)
-
[3]
Karich, A., Kluge, M., Ullrich, R. & Hofrichter, M. Benzene oxygenation and oxidation by the peroxygenase of Agrocybe aegerita.AMB Express3, 5, DOI: 10.1186/2191-0855-3-5 (2013)
-
[4]
Karich, A., Scheibner, K., Ullrich, R. & Hofrichter, M. Exploring the catalase activity of unspecific peroxygenases and the mechanism of peroxide-dependent heme destruction.J. Mol. Catal. B: Enzym.134, 238–246, DOI: 10.1016/j.molcatb. 2016.10.014 (2016)
-
[5]
O., Bormann, S., Hollmann, F., Bloh, J
Burek, B. O., Bormann, S., Hollmann, F., Bloh, J. Z. & Holtmann, D. Hydrogen peroxide driven biocatalysis.Green Chem. 21, 3232–3249, DOI: 10.1039/C9GC00633H (2019)
-
[6]
Hrycay, E. G. & Bandiera, S. M. The monooxygenase, peroxidase, and peroxygenase properties of cytochrome P450.Arch. Biochem. Biophys.522, 71–89, DOI: 10.1016/j.abb.2012.01.003 (2012)
-
[7]
Hofrichter, M.et al.Peroxide-Mediated Oxygenation of Organic Compounds by Fungal Peroxygenases.Antioxidants11, 163, DOI: 10.3390/antiox11010163 (2022)
-
[8]
Antioxidants11, 891, DOI: 10.3390/antiox11050891 (2022)
Linde, D.et al.Structural Characterization of Two Short Unspecific Peroxygenases: Two Different Dimeric Arrangements. Antioxidants11, 891, DOI: 10.3390/antiox11050891 (2022)
-
[9]
Ramirez-Escudero, M.et al.Structural Insights into the Substrate Promiscuity of a Laboratory-Evolved Peroxygenase. ACS Chem. Biol.13, 3259–3268, DOI: 10.1021/acschembio.8b00500 (2018)
-
[10]
Sigmund, M.-C. & Poelarends, G. J. Current state and future perspectives of engineered and artificial peroxygenases for the oxyfunctionalization of organic molecules.Nat. Catal.3, 690–702, DOI: 10.1038/s41929-020-00507-8 (2020)
-
[11]
Costa, G. J. & Liang, R. Understanding the Multifaceted Mechanism of Compound I Formation in Unspecific Peroxygenases through Multiscale Simulations.The J. Phys. Chem. B127, 8809–8824, DOI: 10.1021/acs.jpcb.3c04589 (2023). 12/15 12.Groves, J. T. Using push to get pull.Nat. Chem.6, 89–91, DOI: 10.1038/nchem.1855 (2014)
-
[12]
Green, M. T., Dawson, J. H. & Gray, H. B. Oxoiron(IV) in Chloroperoxidase Compound II Is Basic: Implications for P450 Chemistry.Science304, 1653–1656, DOI: 10.1126/science.1096897 (2004)
-
[13]
Yosca, T. H.et al.Iron(IV)hydroxide pKa and the Role of Thiolate Ligation in C–H Bond Activation by Cytochrome P450. Science342, 825–829, DOI: 10.1126/science.1244373 (2013)
-
[14]
Wang, X., Peter, S., Kinne, M., Hofrichter, M. & Groves, J. T. Detection and Kinetic Characterization of a Highly Reactive Heme–Thiolate Peroxygenase Compound I.J. Am. Chem. Soc.134, 12897–12900, DOI: 10.1021/ja3049223 (2012)
-
[15]
Wang, X., Peter, S., Ullrich, R., Hofrichter, M. & Groves, J. T. Driving Force for Oxygen-Atom Transfer by Heme-Thiolate Enzymes.Angewandte Chemie Int. Ed.52, 9238–9241, DOI: 10.1002/anie.201302137 (2013)
-
[16]
Henkelman, G., Uberuaga, B. P. & Jónsson, H. A climbing image nudged elastic band method for finding saddle points and minimum energy paths.The J. Chem. Phys.113, 9901–9904, DOI: 10.1063/1.1329672 (2000)
-
[17]
Henkelman, G. & Jónsson, H. Improved tangent estimate in the nudged elastic band method for finding minimum energy paths and saddle points.The J. Chem. Phys.113, 9978–9985, DOI: 10.1063/1.1323224 (2000)
-
[18]
Sheppard, D. & Henkelman, G. Paths to which the nudged elastic band converges.J. Comput. Chem.32, 1769–1771, DOI: 10.1002/jcc.21748 (2011)
-
[19]
Taher, M., Dutta Dubey, K. & Mazumdar, S. Computationally guided bioengineering of the active site, substrate access pathway, and water channels of thermostable cytochrome P450, CYP175A1, for catalyzing the alkane hydroxylation reaction.Chem. Sci.14, 14316–14326, DOI: 10.1039/D3SC02857G (2023)
-
[20]
K.et al.Fast product release requires active-site water dynamics in carbonic anhydrase.Nat
Kim, J. K.et al.Fast product release requires active-site water dynamics in carbonic anhydrase.Nat. Commun.16, 4404, DOI: 10.1038/s41467-025-59645-x (2025)
-
[21]
Spin-State Energetics of Heme-Related Models from DFT and Coupled Cluster Calculations.J
Rado´n, M. Spin-State Energetics of Heme-Related Models from DFT and Coupled Cluster Calculations.J. Chem. Theory Comput.10, 2306–2321, DOI: 10.1021/ct500103h (2014)
-
[22]
Schöneboom, J. C. & Thiel, W. The Resting State of P450cam: A QM/MM Study.The J. Phys. Chem. B108, 7468–7478, DOI: 10.1021/jp049596t (2004)
-
[23]
Chen, H., Hirao, H., Derat, E., Schlichting, I. & Shaik, S. Quantum Mechanical/Molecular Mechanical Study on the Mechanisms of Compound I Formation in the Catalytic Cycle of Chloroperoxidase: An Overview on Heme Enzymes.The J. Phys. Chem. B112, 9490–9500, DOI: 10.1021/jp803010f (2008)
-
[24]
Teune, M.et al.Insights into a water-mediated catalytic triad architecture in CE20 carbohydrate esterases.Nat. Commun. 16, 7034, DOI: 10.1038/s41467-025-62387-5 (2025)
-
[25]
Elahi, N. & Zeinalipour-Yazdi, C. D. A DFT assessment of the activation barrier for concerted proton transfer in cyclic water clusters (H2O)n where n = 3–8.Comput. Theor. Chem.1244, 115061, DOI: 10.1016/j.comptc.2024.115061 (2025)
-
[26]
Jacob, C. R. & Reiher, M. Spin in density-functional theory.Int. J. Quantum Chem.112, 3661–3684, DOI: 10.1002/qua. 24309 (2012)
work page doi:10.1002/qua 2012
-
[27]
Li, D., Wang, Y . & Han, K. Recent density functional theory model calculations of drug metabolism by cytochrome P450. Coord. Chem. Rev.256, 1137–1150, DOI: 10.1016/j.ccr.2012.01.016 (2012)
-
[28]
Rev.110, 949–1017, DOI: 10.1021/cr900121s (2010)
Shaik, S.et al.P450 Enzymes: Their Structure, Reactivity, and Selectivity—Modeled by QM/MM Calculations.Chem. Rev.110, 949–1017, DOI: 10.1021/cr900121s (2010)
-
[29]
McIntosh, J. A., Heel, T., Buller, A. R., Chio, L. & Arnold, F. H. Structural Adaptability Facilitates Histidine Heme Ligation in a Cytochrome P450.J. Am. Chem. Soc.137, 13861–13865, DOI: 10.1021/jacs.5b07107 (2015). 31.Unspecific peroxygenase from Collariella virescens: 7zcl, DOI: 10.2210/pdb7zcl/pdb (2022)
-
[30]
Gordon, J. C.et al.H++: a server for estimating p Ka s and adding missing hydrogens to macromolecules.Nucleic Acids Res.33, W368–W371, DOI: 10.1093/nar/gki464 (2005)
-
[31]
Myers, J., Grothaus, G., Narayanan, S. & Onufriev, A. A simple clustering algorithm can be accurate enough for use in calculations of pKs in macromolecules.Proteins: Struct. Funct. Bioinforma.63, 928–938, DOI: 10.1002/prot.20922 (2006)
-
[32]
Anandakrishnan, R., Aguilar, B. & Onufriev, A. V . H++ 3.0: automating pK prediction and the preparation of biomolecular structures for atomistic molecular modeling and simulations.Nucleic Acids Res.40, W537–W541, DOI: 10.1093/nar/gks375 (2012). 13/15 35.GROMACS 2022.2 Source code, DOI: 10.5281/zenodo.6637571 (2022)
-
[33]
Abraham, M. J.et al.GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers.SoftwareX1-2, 19–25, DOI: 10.1016/j.softx.2015.06.001 (2015)
-
[34]
Berendsen, H. J. C., van der Spoel, D. & van Drunen, R. GROMACS: A message-passing parallel molecular dynamics implementation.Comput. Phys. Commun.91, 43–56, DOI: 10.1016/0010-4655(95)00042-E (1995)
-
[35]
Hess, B., Kutzner, C., van der Spoel, D. & Lindahl, E. GROMACS 4: Algorithms for Highly Efficient, Load-Balanced, and Scalable Molecular Simulation.J. Chem. Theory Comput.4, 435–447, DOI: 10.1021/ct700301q (2008)
-
[36]
Lindahl, E., Hess, B. & van der Spoel, D. GROMACS 3.0: a package for molecular simulation and trajectory analysis. Mol. modeling annual7, 306–317, DOI: 10.1007/s008940100045 (2001)
-
[37]
Páll, S., Abraham, M. J., Kutzner, C., Hess, B. & Lindahl, E. Tackling Exascale Software Challenges in Molecular Dynamics Simulations with GROMACS. In Markidis, S. & Laure, E. (eds.)Solving Software Challenges for Exascale, 3–27, DOI: 10.1007/978-3-319-15976-8_1 (Springer International Publishing, Cham, 2015)
-
[38]
Páll, S.et al.Heterogeneous parallelization and acceleration of molecular dynamics simulations in GROMACS.The J. Chem. Phys.153, 134110, DOI: 10.1063/5.0018516 (2020)
-
[39]
Bioinformatics29, 845–854, DOI: 10.1093/bioinformatics/btt055 (2013)
Pronk, S.et al.GROMACS 4.5: a high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics29, 845–854, DOI: 10.1093/bioinformatics/btt055 (2013)
-
[40]
Van Der Spoel, D.et al.GROMACS: Fast, flexible, and free.J. Comput. Chem.26, 1701–1718, DOI: 10.1002/jcc.20291 (2005)
-
[41]
Soteras Gutiérrez, I.et al.Parametrization of halogen bonds in the CHARMM general force field: Improved treatment of ligand–protein interactions.Bioorganic & Medicinal Chem.24, 4812–4825, DOI: 10.1016/j.bmc.2016.06.034 (2016)
-
[42]
Vanommeslaeghe, K.et al.CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields.J. Comput. Chem.31, 671–690, DOI: 10.1002/jcc.21367 (2010)
-
[43]
Vanommeslaeghe, K. & MacKerell, A. D. J. Automation of the CHARMM General Force Field (CGenFF) I: Bond Perception and Atom Typing.J. Chem. Inf. Model.52, 3144–3154, DOI: 10.1021/ci300363c (2012). 47.Vanommeslaeghe, K., Raman, E. P. & MacKerell, A. D. J. Automation of the CHARMM General Force Field (CGenFF) II: Assignment of Bonded Parameters and Partial A...
-
[44]
Yu, W., He, X., Vanommeslaeghe, K. & MacKerell Jr., A. D. Extension of the CHARMM general force field to sulfonyl-containing compounds and its utility in biomolecular simulations.J. Comput. Chem.33, 2451–2468, DOI: 10.1002/jcc.23067 (2012)
-
[45]
Lemkul, J. From proteins to perturbed hamiltonians: A suite of tutorials for the GROMACS-2018 molecular simulation package.Living J. Comput. Mol. Sci.1, DOI: 10.33011/livecoms.1.1.5068 (2019). 50.Schrödinger, LLC & DeLano, W. The PyMOL molecular graphics system, version 1.8 (2015)
-
[46]
D., de Oliveira, C
Durrant, J. D., de Oliveira, C. A. F. & McCammon, J. A. POVME: An Algorithm for Measuring Binding-Pocket V olumes. J. molecular graphics & modelling29, 773–776 (2011)
2011
-
[47]
Durrant, J. D., V otapka, L., Sørensen, J. & Amaro, R. E. POVME 2.0: An Enhanced Tool for Determining Pocket Shape and V olume Characteristics.J. Chem. Theory Comput.10, 5047–5056, DOI: 10.1021/ct500381c (2014)
-
[48]
Model Simul Mater Sci Eng 18:015012
Stukowski, A. Visualization and analysis of atomistic simulation data with OVITO–the Open Visualization Tool.Model. Simul. Mater. Sci. Eng.18, 015012, DOI: 10.1088/0965-0393/18/1/015012 (2009)
-
[49]
Trott, O. & Olson, A. J. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading.J. Comput. Chem.31, 455–461, DOI: 10.1002/jcc.21334 (2010)
-
[50]
Eberhardt, J., Santos-Martins, D., Tillack, A. F. & Forli, S. AutoDock Vina 1.2.0: New Docking Methods, Expanded Force Field, and Python Bindings.J. Chem. Inf. Model.61, 3891–3898, DOI: 10.1021/acs.jcim.1c00203 (2021)
-
[51]
Senn, H. M. & Thiel, W. QM/MM methods for biological systems.At. Approaches Mod. Biol.268, 173–290, DOI: 10.1007/128_2006_084 (2006). 57.Neese, F. The ORCA program system.WIREs Comput. Mol. Sci.2, 73–78, DOI: 10.1002/wcms.81 (2012)
-
[52]
Software Update: The ORCA Program System—Version 6.0.WIREs Comput
Neese, F. Software Update: The ORCA Program System—Version 6.0.WIREs Comput. Mol. Sci.15, e70019, DOI: 10.1002/wcms.70019 (2025). 14/15
-
[53]
& Wennmohs, F.ORCA 6.0 Manual
Neese, F. & Wennmohs, F.ORCA 6.0 Manual. FACCTs GmbH, Mülheim a. d. Ruhr, Germany, 1st edn. (2025). Available at https://www.faccts.de/docs/orca/6.0/manual/index.html. Acknowledgments (not compulsory) C.J. and T.J. gratefully acknowledge funding through the DFG (CRC 1316-2). The authors acknowledge support by the state of Baden-Württemberg through bwHPC a...
2025
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.