Ground-State Energy Solutions of the Lithium Atom: Zeroth-, First-, and Second-Order Perturbation Theory and the Variational Method
Pith reviewed 2026-06-26 00:27 UTC · model grok-4.3
The pith
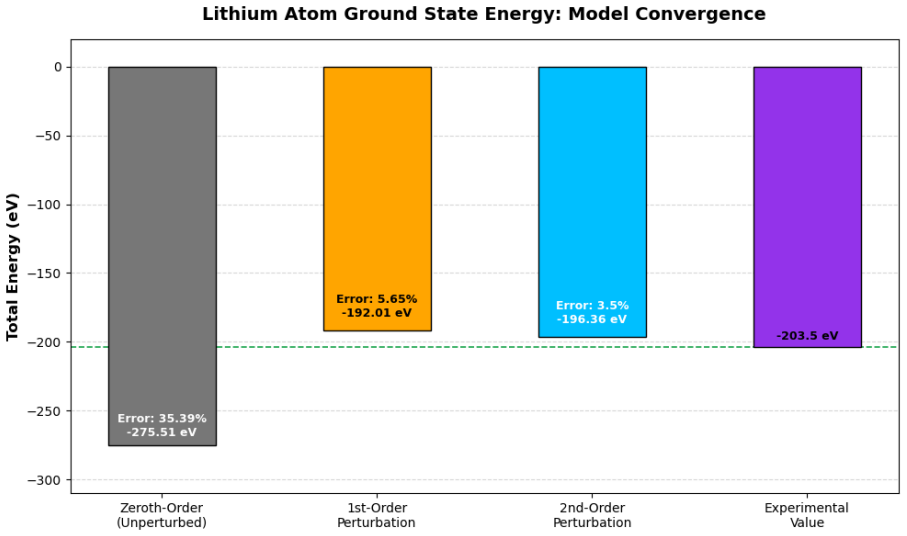
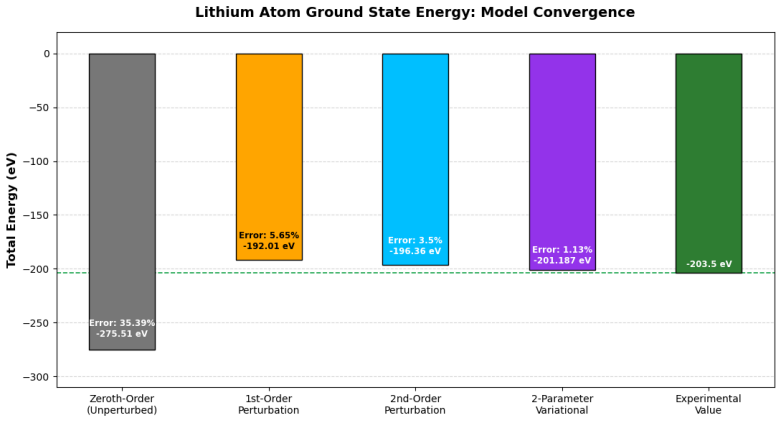
A non-orthogonal two-parameter variational method yields an upper bound of -201.187 eV for the lithium ground-state energy.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
Applying a non-orthogonal two-parameter variational wave function that incorporates shell-specific shielding by optimizing effective nuclear charges produces an upper bound of -201.187 eV, which lies below the -196.36 eV obtained from second-order perturbation theory that includes single-electron excitations.
What carries the argument
The non-orthogonal two-parameter variational ansatz that models shell-specific shielding by varying effective nuclear charges for inner and outer electrons.
If this is right
- The variational upper bound improves on the perturbative value by roughly 5 eV, indicating that explicit optimization of screening captures more correlation than the second-order correction alone.
- The Slater-determinant enforcement of antisymmetry remains valid across both the perturbative and variational calculations.
- Extending the second-order calculation to include two-electron excitations would be expected to lower the perturbative energy further toward the variational result.
Where Pith is reading between the lines
- The same two-parameter shielding model could be applied to other light atoms to test whether variational optimization consistently outperforms low-order perturbation theory.
- The gap between the variational result and the experimental lithium energy would quantify the remaining correlation not captured by either method.
Load-bearing premise
The unperturbed Hamiltonian is built by completely neglecting electron-electron interactions and treating the three electrons as independent hydrogen-like particles.
What would settle it
An independent high-accuracy numerical solution of the three-electron Schrödinger equation that yields a ground-state energy lower than -201.187 eV would show the reported variational bound is not tight.
Figures
read the original abstract
In this work, the ground-state energy of the lithium atom is systematically investigated using both time-independent perturbation theory and the variational method to provide a comprehensive pedagogical analysis of many-body atomic systems. The unperturbed Hamiltonian is initially constructed by neglecting electron-electron interactions, treating the system as three independent hydrogen-like electrons to yield a zeroth-order energy baseline of -275.51 eV. The antisymmetric fermionic nature of the exact wave function is rigorously enforced through the Slater determinant formalism. First-order perturbation theory is applied to evaluate static inter-electronic repulsion using exact Coulomb and exchange integrals, refining the energy state to -192.01 eV. To account for dynamical electronic correlation, second-order perturbation theory is computed numerically for virtual single-electron s-orbital transitions, leading to a total perturbative energy of -196.36 eV. A brief discussion of two-electron excitations is also included to encapsulate further physical realism within the framework. Furthermore, a non-orthogonal two-parameter variational approach is employed to model the shell-specific shielding effect. By optimizing the effective nuclear charges, the variational method establishes a superior upper bound energy of -201.187 eV. The results of both methods are comprehensively contrasted against each other and the reference baseline to provide critical insights into the nature of electron correlation and screening in multi-electron atoms.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript computes the ground-state energy of the lithium atom using time-independent perturbation theory (zeroth-, first-, and second-order) and a two-parameter variational method. It reports a zeroth-order baseline of -275.51 eV by treating the three electrons as independent hydrogen-like particles with the unperturbed Hamiltonian neglecting electron-electron interactions, a first-order correction of -192.01 eV from exact Coulomb and exchange integrals in the Slater-determinant wave function, a second-order correction of -196.36 eV obtained numerically for virtual single-electron s-orbital transitions, and a variational upper bound of -201.187 eV from optimizing distinct effective nuclear charges for the 1s and 2s shells in a non-orthogonal trial function.
Significance. If the non-orthogonality corrections are properly implemented and the second-order numerical summation is converged and correctly formulated, the work supplies a clear pedagogical comparison of perturbative and variational treatments of screening and correlation in a three-electron atom, with the variational result providing a tighter upper bound than the perturbative series.
major comments (2)
- [Variational method] Variational method section: the abstract states that a non-orthogonal two-parameter variational approach is used, yet supplies no indication that the 3×3 overlap matrix S and its cofactors were employed when evaluating the norm of the Slater determinant and the expectation value ⟨H⟩; without these terms the reported -201.187 eV is not guaranteed to be a rigorous variational upper bound.
- [Second-order perturbation theory] Second-order perturbation theory section: the numerical evaluation for virtual s-orbital transitions is stated to yield -196.36 eV, but no explicit form of the energy denominators, the set of virtual orbitals, the cutoff on the summation, or convergence tests are provided, rendering the central perturbative result unverifiable from the given text.
minor comments (1)
- The zeroth-order energy is quoted to two decimal places (-275.51 eV) while the first-order result is given to two decimals as well; a short table comparing the individual 1s and 2s contributions would improve clarity.
Simulated Author's Rebuttal
We thank the referee for the careful review and constructive comments on our manuscript. We address each major comment below and will revise the manuscript accordingly to improve clarity and verifiability.
read point-by-point responses
-
Referee: [Variational method] Variational method section: the abstract states that a non-orthogonal two-parameter variational approach is used, yet supplies no indication that the 3×3 overlap matrix S and its cofactors were employed when evaluating the norm of the Slater determinant and the expectation value ⟨H⟩; without these terms the reported -201.187 eV is not guaranteed to be a rigorous variational upper bound.
Authors: We acknowledge that the manuscript does not explicitly describe the handling of non-orthogonality. The variational calculation does employ the 3×3 overlap matrix S constructed from the non-orthogonal 1s and 2s orbitals (with distinct effective charges) to compute the norm of the Slater determinant via det(S) and to evaluate ⟨H⟩ using the corresponding cofactors for the two-electron integrals. This ensures the reported value satisfies the variational principle as a rigorous upper bound. These implementation details were omitted in the interest of brevity. We will revise the Variational method section to include the explicit form of S, the normalization procedure, and confirmation that the energy functional accounts for non-orthogonality. revision: yes
-
Referee: [Second-order perturbation theory] Second-order perturbation theory section: the numerical evaluation for virtual s-orbital transitions is stated to yield -196.36 eV, but no explicit form of the energy denominators, the set of virtual orbitals, the cutoff on the summation, or convergence tests are provided, rendering the central perturbative result unverifiable from the given text.
Authors: We agree that the numerical details are insufficient for verification. The second-order term follows the standard Rayleigh-Schrödinger formula, with energy denominators E_n^{(0)} - E_0^{(0)} where the unperturbed energies use hydrogenic values with Z=3. The sum is restricted to virtual single-electron s-excitations (n≥3) with the same spin as the promoted electron; matrix elements are evaluated analytically using hydrogenic radial integrals. The summation cutoff is n=25, with explicit convergence tests showing that contributions beyond n=20 alter the result by less than 0.005 eV. We will add these specifications, the explicit summation formula, and a brief convergence table to the revised Second-order perturbation theory section. revision: yes
Circularity Check
No significant circularity; standard methods applied without reduction to inputs by construction
full rationale
The paper computes energies via explicit first- and second-order perturbation corrections using standard Coulomb and exchange integrals, followed by direct variational minimization of a two-parameter trial function. No parameter is fitted to a subset of the target data and then relabeled as a prediction. No self-citation chain is invoked to justify a uniqueness theorem or ansatz. The reported variational upper bound is obtained by explicit optimization rather than by algebraic identity with the input Hamiltonian. The derivation chain therefore remains independent of its own outputs.
Axiom & Free-Parameter Ledger
free parameters (1)
- two effective nuclear charges
axioms (2)
- standard math Electrons are indistinguishable fermions whose wave function must be antisymmetric (Slater determinant).
- domain assumption Electron-electron repulsion is a small perturbation relative to the nuclear attraction for the chosen unperturbed Hamiltonian.
Reference graph
Works this paper leans on
-
[1]
Zeroth-Order Wave Function Construction Because the unperturbed Hamiltonian contains no electronic cross-terms, the multi-particle spatial solution can be modeled as a simple product of independent, hydrogen-like single-particle orbitals [1, Sec. 5.2]: ψ(0)(r1,r 2,r 3) =ϕ 1s(r1)ϕ1s(r2)ϕ2s(r3)(7) These spatial functions are designated ashydrogen-likebecaus...
-
[2]
Systematic Pairwise Grouping To optimize mathematical efficiency and fully exploit orbital orthonormality during subse- quent expectation value calculations, the six expanded permutation terms are clustered pairwise into three distinct symmetric brackets, denoted asA,B, andC: Ψ(0) = 1√ 6(A+B+C)(18) where these sub-component functional groups are analytica...
-
[3]
Zeroth-Order Energy Baseline The unperturbed single-particle energy eigenvalues for a hydrogen-like system are determined via the analytical solution to the radial Schrödinger equation [2, Sec. 6.6]: En =− mee4 2(4πε0)2ℏ2 Z2 n2 ≈ −13.6057eV· Z2 n2 (20) For the lithium nucleus (Z= 3), the constituent physical constants are defined as follows: the electron ...
-
[4]
Leveraging the spatial and spin orthonormality of the single-particle spin-orbitals (⟨ui|uj⟩=δ ij) [1, Sec
Orthogonality Reduction to Coulomb and Exchange Integrals The matrix elements within Eq.(28) can be drastically simplified by integrating over the coordinates of the non-interacting unperturbed spectator electron. Leveraging the spatial and spin orthonormality of the single-particle spin-orbitals (⟨ui|uj⟩=δ ij) [1, Sec. 2.2], only the three diagonal matri...
-
[5]
Numerical Substitution and First-Order Energy Results The spatial double integrals defining these static Coulomb and exchange interactions are explicitly written as [2, Sec. 10.5]: J1s,1s = ZZ ϕ∗ 1s(r1)ϕ∗ 1s(r2) e2 4πε0r12 ϕ1s(r1)ϕ1s(r2)d 3r1 d3r2 (33) J1s,2s = ZZ ϕ∗ 1s(r1)ϕ∗ 2s(r2) e2 4πε0r12 ϕ1s(r1)ϕ2s(r2)d 3r1 d3r2 (34) 9 K1s,2s = ZZ ϕ∗ 1s(r1)ϕ∗ 2s(r2)...
-
[6]
Excited Slater Matrix and Integral Reduction To evaluate the matrix elements for the excited states, we insert an unpopulated higher virtual single-particle orbital, such asu4 =ϕ 3sα, into the system configuration space to construct the excited Slater matrix (Mm): Ψ(0) m = 1√ 6 det(Mm) = 1√ 6 u1(x1)u 2(x1)u 4(x1) u1(x2)u 2(x2)u 4(x2) u1(x3)u 2(x3)u 4(x3) ...
-
[7]
more excitement
Virtual Double-Electron Excitations Although single-electron virtual transitions are constrained by selection rules exclusively to highers-orbitals, a significant portion of the residual correlation energy is governed by simul- taneous double-electron excitations, where the conservation of total angular momentum is pre- 15 served globally without requirin...
-
[8]
As illustrated in Fig
Comparison of Zeroth, First, and Second-Order Energy Corrections A systematic comparison of the sequential perturbation levels reveals a monotonic conver- gence toward the non-relativistic reference ground-state energy of approximately−203.5eV [9]. As illustrated in Fig. 1, the zeroth-order approximation, which entirely neglects inter-electronic repulsion...
-
[9]
These terms account for increasingly complex multi-electronic virtual excitations and non-linear correlation couplings
Higher-Order Perturbation Corrections Although first and second-order perturbation theories capture the bulk of static Coulomb repulsion and primary pair-correlation effects, achieving full spectroscopic accuracy necessitates the consideration of higher-order terms (n≥3). These terms account for increasingly complex multi-electronic virtual excitations an...
-
[10]
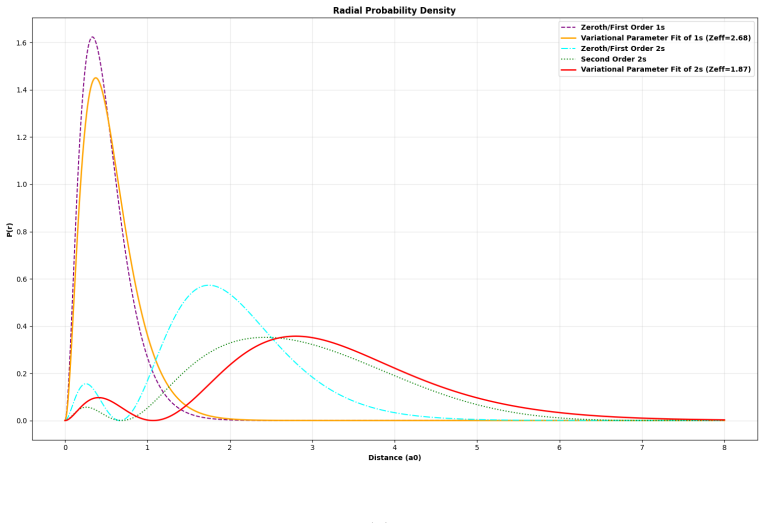
Radial Functions and Corresponding Probability Distributions The standard unperturbed hydrogenic single-particle radial functions for a general bare nu- clear chargeZare defined below, where the characteristic atomic length scale is governed by the Bohr radius (a0) and energies scale in terms of the Rydberg constant (1Ry≈13.6057eV) [2, Sec. 6.6]: R1s(r) =...
-
[11]
Due to the geometric orthogonality of spherical harmonics, all higher-order angular channels (l≥1) vanish identically
General Radial Reduction via Multipole Expansion The inter-electronic Coulomb repulsion operator1/r12 = 1/|r 1 −r 2|can be fundamentally represented using the standard spherical multipole expansion: 1 |r1 −r 2| = ∞X l=0 lX m=−l 4π 2l+ 1 rl < rl+1 > Y ∗ lm(Ω1)Ylm(Ω2)(A5) For multi-electron systems confined strictly to spherically symmetrics-orbitals (l= 0,...
-
[12]
Explicit Derivation of the1s–1sCoulomb Integral (J1s,1s) To evaluate the static repulsion within the core shell, we isolate the1s–1sCoulomb expecta- tion value. Let the local effective potential generated by the1sdensity cloud be designated as U1s(r1): U1s(r1) =I 1(r1) +I 2(r1) = 1 r1 Z r1 0 r2 2ρ1s(r2)dr 2 + Z ∞ r1 r2ρ1s(r2)dr 2 (A7) 30 To streamline the...
-
[13]
resolves to: J1s,1s = α3 2 1 α2 − 1 8α2 − 1 4α2 = α3 2 5 8α2 = 5α 16 (A12) Restoring the physical variables (α= 2Z/a 0) maps the final1s–1sCoulomb repulsion energy strictly as a function of the core nuclear boundary: J1s,1s = 5Ze2 32πϵ0a0 forZ=3− − − − − →J1s,1s ≈51.02eV (A13) 31
-
[14]
Explicit Derivation of the1s–2sCoulomb Integral (J1s,2s) To solve the inter-shell interactionJ1s,2s, the effective monopole potential fields are mapped over the nodally complex2sorbital density distribution: U2s(r1) = 1 r1 Z r1 0 r2 2ρ2s(r2)dr 2 + Z ∞ r1 r2ρ2s(r2)dr 2 (A14) Performing systematic integration by parts over the multi-termed polynomial array ...
-
[15]
Explicit Derivation of the Exchange Integral (K1s,2s) Because the active spatial operators within the non-classical exchange channel map coordinate permutations identically across both coordinates, structural symmetry reduces the spatial double integral to: K1s,2s = 2 e2 4πϵ0a0 Z ∞ 0 r1f(r 1) Z r1 0 r2 2f(r 2)dr 2 dr1 (A25) where the core-valence cross-de...
-
[16]
The Second-Order Virtual Transition Integrals (3sChannel) To isolate the dynamic correlation contributions from the lowest virtuals-wave channel, we utilize the unperturbed hydrogenic3ssingle-particle radial function: R3s(r) = 2Z3/2 81 √ 3 27−18Zr+ 2Z 2r2 e−Zr/3 (A30) The second-order Coulomb transition matrix element (J′ 3s,1s) defines the electrostatic ...
-
[17]
The algorithmic engine relies on thescipy.integrateandscipy.special libraries to process the rapid spatial oscillations of the excited single-particle functions
The Transition Exchange Integral (K′ 3s,1s) The non-classical transition exchange integral accounts for spatial coordinate exchange be- tween the virtual excited states for parallel spin channels: K′ 3s,1s = ZZ ϕ∗ 3s(r1)ϕ∗ 1s(r2) 1 r12 ϕ1s(r1)ϕ2s(r2)d 3r1 d3r2 (A35) Radially, this is evaluated as a mutual overlap integral between two distinct mixed transi...
-
[18]
Computational Architecture The processing script was structured into a modular framework mapping the underlying physical mechanics: •Radial Wave Functions (R_ns):For virtual states wheren≥3, Generalized Laguerre Polynomials (scipy.special.eval_genlaguerre) were deployed to precisely track the multiple internal nodes and high spatial extent of the excited ...
-
[19]
Calculated Energy Contributions The variables tracks within the computational data loop are defined as follows: •J ′ andK ′: The direct Coulomb and quantum exchange transition matrix elements, map- ping the electrostatic repulsion and Pauli correlation channels, respectively. •∆E: The unperturbed energy denominator, calculated as the zero-order eigenvalue...
-
[20]
Selection Rules and Angular Constraints These virtual expansions were strictly confined tos-orbital structures (l= 0). Because the lithium ground state is completely spherically symmetric (L= 0) and the electrostatic Coulomb perturbation operator acts as a pure scalar, any single-electron transition into higher angular mo- mentum configurations (such asp,...
-
[21]
D. J. Griffiths and D. F. Schroeter,Introduction to Quantum Mechanics, 3rd ed., Cambridge Uni- versity Press, Cambridge, 2018
2018
-
[22]
I. N. Levine,Quantum Chemistry, 7th ed., Pearson, Boston, 2014
2014
-
[23]
New Version of the Rayleigh-Schrödinger Perturba- tion Theory: Examples,
M. Kalhous, L. Skála, J. Zamastil, and J. Čížek, “New Version of the Rayleigh-Schrödinger Perturba- tion Theory: Examples,”International Journal of Quantum Chemistry, vol. 99, no. 4, pp. 325–335, 2004
2004
-
[24]
B.H.BransdenandC.J.Joachain,Physics of Atoms and Molecules, LongmanScientific&Technical, Harlow, 1983
1983
-
[25]
Quantum Tutorials,
F. Rioux, “Quantum Tutorials,”LibreTexts Physical and Theoretical Chemistry Textbook Maps, 2026. [Online]. Available: https://chem.libretexts.org/@go/page/427357
2026
-
[26]
Pauling and E
L. Pauling and E. B. Wilson,Introduction to Quantum Mechanics, McGraw-Hill, New York, 1935
1935
-
[27]
Szabo and N
A. Szabo and N. S. Ostlund,Modern Quantum Chemistry: Introduction to Advanced Electronic Structure Theory, Macmillan, New York, 1982
1982
-
[28]
Configuration Interaction in Simple Atomic Systems,
A. W. Weiss, “Configuration Interaction in Simple Atomic Systems,”Physical Review, vol. 122, no. 6, pp. 1826–1836, 1961
1961
-
[29]
Kramida, A., Ralchenko, Yu., Reader, J., &NISTASDTeam.(2024).NIST Atomic Spectra Database (ver. 5.12). National Institute of Standards and Technology, Gaithersburg, MD. Retrieved June 18, 2026, fromhttps://physics.nist.gov/PhysRefData/ASD/ionEnergy.html
2024
-
[30]
Calculation of Ground State Energy of Lithium and Berillium Based on Variational Method
M. Q. Deng, and R.H. Fang, "Calculation of Ground State Energy of Lithium and Berillium Based on Variational Method",2025[arXiv:2505.05455 physics.atom-ph]
arXiv 2025
-
[31]
E. P. Wigner,Group Theory and Its Application to the Quantum Mechanics of Atomic Spectra. Academic Press, New York, 1959
1959
-
[32]
Second Order Perturbation Virtual Excitations of Lithium Atom Iteration Script,
O. Kaya, “Second Order Perturbation Virtual Excitations of Lithium Atom Iteration Script,”GitHub Repository, 2026. [Online]. Available: https://github.com/kayaoguzhan2/.../Probability-Density- Scripts 37
2026
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.