CoTAR: Topology and Atomic State Reconstruction in Condensed Phases
Pith reviewed 2026-06-29 00:41 UTC · model grok-4.3
The pith
CoTAR reconstructs molecular topologies, formal charges, and unpaired electrons from atomic species, coordinates, and total charge using a hybrid GNN-HMM framework.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
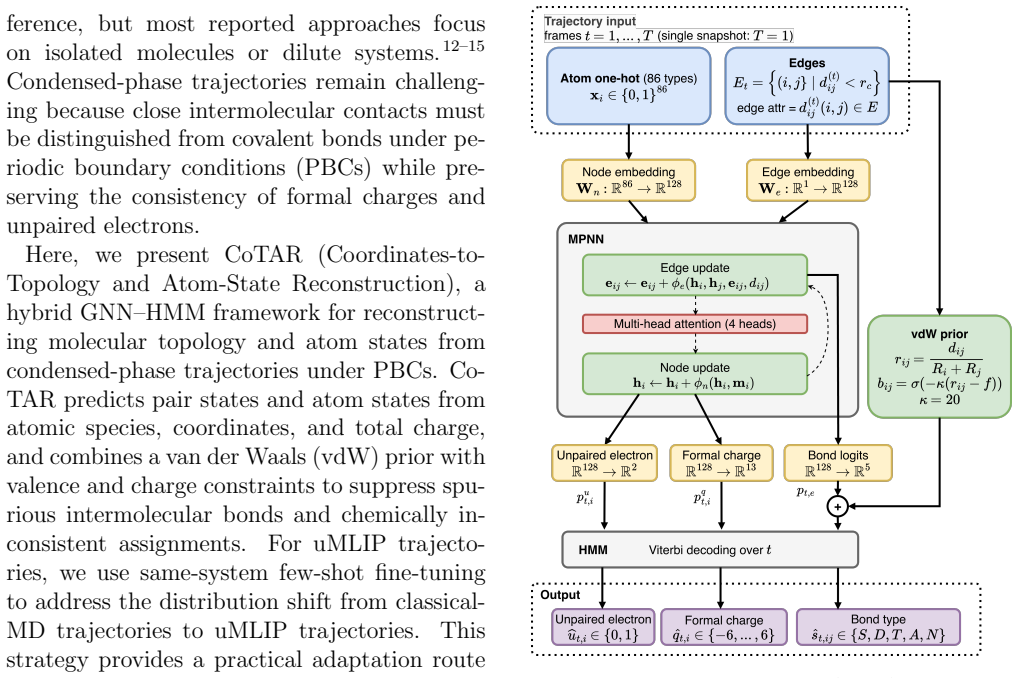
CoTAR is a hybrid graph neural network and hidden Markov model that reconstructs molecular topology, formal charges, and unpaired electrons by message passing on a proximity graph augmented by a van der Waals prior and chemical constraints, followed by temporal smoothing; the framework yields a bond-order-weighted F1 score of 0.906 across 128 nonreactive condensed-phase systems on classical MD data and raises the fraction of valid uMLIP snapshots from 38.6 percent to 84.7 percent after few-shot fine-tuning.
What carries the argument
The CoTAR hybrid GNN-HMM framework that performs message passing on proximity graphs together with a van der Waals prior, chemical constraints, and temporal smoothing.
If this is right
- Reconstructed topologies enable bond-aware analysis of uMLIP trajectories.
- Few-shot fine-tuning raises the valid-snapshot rate on uMLIP data from 38.6 percent to 84.7 percent.
- The topologies support downstream classical MD simulations.
- HMM smoothing increases system-level MD simulation feasibility from 83.6 percent to 85.9 percent.
Where Pith is reading between the lines
- The method could link uMLIP dynamics directly to existing classical force-field pipelines without manual topology assignment.
- Relaxing the nonreactive assumption might allow the same reconstruction machinery to handle bond-breaking events.
- Analogous proximity-graph plus constraint models could be tested on other particle simulations that lack explicit connectivity.
Load-bearing premise
The 128 tested nonreactive systems represent the condensed-phase cases where uMLIP trajectories are used, and the combination of proximity-graph message passing, van der Waals prior, and chemical constraints produces chemically valid topologies without further system-specific tuning.
What would settle it
Applying CoTAR to uMLIP trajectories from a condensed-phase system outside the original 128 and observing that few-shot fine-tuning leaves the valid-snapshot rate near 38.6 percent would show the reconstruction does not generalize.
Figures
read the original abstract
Universal machine learning interatomic potentials (uMLIPs) enable condensed-phase molecular dynamics (MD) simulations with near-first-principles accuracy, but their lack of explicit molecular topology limits bond-aware analysis and reconnection to classical force fields. Here, we present CoTAR, a hybrid graph neural network (GNN)--hidden Markov model (HMM) framework that reconstructs molecular topology, formal charges, and unpaired electrons from atomic species, coordinates, and total charge by combining message passing on a proximity graph with a van der Waals prior, chemical constraints, and temporal smoothing. Across 128 nonreactive, topology-preserving condensed-phase systems, CoTAR achieved a bond-order-weighted F1 score of 0.906 on classical-MD data; for uMLIP trajectories, few-shot fine-tuning improved the valid-snapshot rate from 38.6\% to 84.7\%. The reconstructed topologies also supported downstream classical MD simulations, and HMM smoothing improved system-level MD simulation feasibility from 83.6\% to 85.9\%, indicating that CoTAR provides a practical framework for bond-aware analysis of condensed-phase uMLIP trajectories.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript introduces CoTAR, a hybrid GNN-HMM framework that reconstructs molecular topology, formal charges, and unpaired electrons from atomic species, coordinates, and total charge by combining message passing on a proximity graph with a van der Waals prior, chemical constraints, and temporal smoothing. Across 128 nonreactive, topology-preserving condensed-phase systems, it reports a bond-order-weighted F1 score of 0.906 on classical-MD data; few-shot fine-tuning on uMLIP trajectories improves the valid-snapshot rate from 38.6% to 84.7%. The reconstructed topologies support downstream classical MD simulations, and HMM smoothing raises system-level MD feasibility from 83.6% to 85.9%.
Significance. If the reported metrics are robust, CoTAR would address a practical gap in uMLIP usage by enabling bond-aware analysis and reconnection to classical force fields without system-specific tuning. The scale of testing (128 systems) and the quantified improvement in valid snapshots constitute a concrete contribution to the field.
minor comments (3)
- [Abstract] Abstract: the dataset composition, selection criteria, and diversity metrics for the 128 systems are not described, which would help readers assess representativeness of the tested condensed-phase cases.
- [Abstract] Abstract: performance numbers are given without error bars, standard deviations, or details on train/test splits and ablation studies; adding these would strengthen the presentation of the F1 and valid-snapshot results.
- The manuscript would benefit from a brief comparison table or section contrasting CoTAR against existing topology-reconstruction methods (e.g., rule-based or other GNN approaches) to clarify the incremental advance.
Simulated Author's Rebuttal
We thank the referee for their positive assessment of CoTAR, the recognition of its practical utility for uMLIP trajectories, and the recommendation for minor revision. No specific major comments were provided in the report.
Circularity Check
No significant circularity
full rationale
The abstract and available description present CoTAR as a hybrid GNN-HMM method whose performance metrics (bond-order-weighted F1 of 0.906 on 128 systems; valid-snapshot rate improvement from 38.6% to 84.7% after few-shot fine-tuning) are reported as direct empirical measurements on classical-MD and uMLIP trajectories. No equations, parameter-fitting steps, or self-citations appear in the supplied text that would reduce any claimed prediction or reconstruction result to a tautology by construction. The method description (proximity-graph message passing plus van der Waals prior plus chemical constraints plus temporal smoothing) is stated at a level that does not exhibit self-definitional, fitted-input, or self-citation-load-bearing circularity. The evaluation is therefore self-contained against external benchmarks.
Axiom & Free-Parameter Ledger
axioms (1)
- domain assumption Message passing on a proximity graph combined with HMM temporal smoothing can recover chemically valid topologies when supplemented by a van der Waals prior and chemical constraints.
Reference graph
Works this paper leans on
-
[1]
Accelerated ReaxFF Simulations for Describing the Reactive Cross-Linking of Polymers
Vashisth, Aniruddh and Ashraf, Chowdhury and Zhang, Weiwei and Bakis, Charles E and van Duin, Adri C T. Accelerated ReaxFF Simulations for Describing the Reactive Cross-Linking of Polymers. J. Phys. Chem. A
-
[2]
Simulations of the biodegradation of citrate-based polymers for artificial scaffolds using accelerated reactive molecular dynamics
Dasgupta, Nabankur and Yilmaz, Dundar E and van Duin, Adri. Simulations of the biodegradation of citrate-based polymers for artificial scaffolds using accelerated reactive molecular dynamics. J. Phys. Chem. B
-
[3]
ReaxFF : A Reactive Force Field for Hydrocarbons
van Duin, Adri C T and Dasgupta, Siddharth and Lorant, Francois and Goddard, William A. ReaxFF : A Reactive Force Field for Hydrocarbons. J. Phys. Chem. A
-
[4]
Escaping free-energy minima
Laio, Alessandro and Parrinello, Michele. Escaping free-energy minima. Proc. Natl. Acad. Sci. U. S. A
-
[5]
Ilyes Batatia and David Peter Kovacs and Gregor N. C. Simm and Christoph Ortner and Gabor Csanyi , booktitle=. 2022 , url=
2022
-
[6]
The Design Space of E(3)-Equivariant Atom-Centered Interatomic Potentials , author =. 2022 , number =. doi:10.48550/arXiv.2205.06643 , archiveprefix =. 2205.06643 , eprinttype =
-
[7]
CHGNet as a pretrained universal neural network potential for charge-informed atomistic modelling
Deng, Bowen and Zhong, Peichen and Jun, Kyujung and Riebesell, Janosh and Han, Kevin and Bartel, Christopher J and Ceder, Gerbrand. CHGNet as a pretrained universal neural network potential for charge-informed atomistic modelling. Nature Machine Intelligence
-
[8]
Towards universal neural network potential for material discovery applicable to arbitrary combination of 45 elements
Takamoto, So and Shinagawa, Chikashi and Motoki, Daisuke and Nakago, Kosuke and Li, Wenwen and Kurata, Iori and Watanabe, Taku and Yayama, Yoshihiro and Iriguchi, Hiroki and Asano, Yusuke and Onodera, Tasuku and Ishii, Takafumi and Kudo, Takao and Ono, Hideki and Sawada, Ryohto and Ishitani, Ryuichiro and Ong, Marc and Yamaguchi, Taiki and Kataoka, Toshik...
-
[9]
A universal graph deep learning interatomic potential for the periodic table
Chen, Chi and Ong, Shyue Ping. A universal graph deep learning interatomic potential for the periodic table. Nat. Comput. Sci
-
[10]
The potential of neural network potentials
Duignan, Timothy T. The potential of neural network potentials. ACS Phys. Chem. Au
-
[11]
Neural network potentials for chemistry: concepts, applications and prospects
Käser, Silvan and Vazquez-Salazar, Luis Itza and Meuwly, Markus and Töpfer, Kai. Neural network potentials for chemistry: concepts, applications and prospects. Digit. Discov
-
[12]
An overview about neural networks potentials in molecular dynamics simulation
Martin-Barrios, Raidel and Navas-Conyedo, Edisel and Zhang, Xuyi and Chen, Yunwei and Gulín-González, Jorge. An overview about neural networks potentials in molecular dynamics simulation. Int. J. Quantum Chem
-
[13]
Simulations of glass transition and mechanical behavior of off-stoichiometric crosslinked polymers
Bezik, Cody T and Redline, Erica M and Foster, Jeffrey C and Frischknecht, Amalie L. Simulations of glass transition and mechanical behavior of off-stoichiometric crosslinked polymers. Macromolecules
-
[14]
Verification for Temperature Dependence of Tacticity in Polystyrene Radical Polymerization with the Combination of Reaction Pathway Analysis and Red Moon Methodology
Rao, Zizhen and Takayanagi, Masayoshi and Nagaoka, Masataka. Verification for Temperature Dependence of Tacticity in Polystyrene Radical Polymerization with the Combination of Reaction Pathway Analysis and Red Moon Methodology. J. Phys. Chem. B
-
[15]
Molecular modeling of reactive systems with REACTER
Gissinger, Jacob R and Jensen, Benjamin D and Wise, Kristopher E. Molecular modeling of reactive systems with REACTER. Comput. Phys. Commun
-
[16]
Enhancing epoxy resin curing: Investigating the catalytic role of water as a trace impurity in dense crosslinked network formation using an advanced cat- GRRM / MC / MD Method1
Xi, Yingxiao and Fukuzawa, Hironobu and Kikugawa, Gota and Zhao, Yinbo and Kawagoe, Yoshiaki and Okabe, Tomonaga and Kishi, Hajime and Kishimoto, Naoki. Enhancing epoxy resin curing: Investigating the catalytic role of water as a trace impurity in dense crosslinked network formation using an advanced cat- GRRM / MC / MD Method1. Polymer (Guildf.)
-
[17]
Development of cat- GRRM / MC / MD method for the simulation of cross-linked network structure formation with molecular autocatalysis
Xi, Yingxiao and Fukuzawa, Hironobu and Fukunaga, Shoji and Kikugawa, Gota and Zhao, Yinbo and Kawagoe, Yoshiaki and Okabe, Tomonaga and Kishimoto, Naoki. Development of cat- GRRM / MC / MD method for the simulation of cross-linked network structure formation with molecular autocatalysis. Molecular Catalysis
-
[18]
Curing reaction of epoxy resin composed of mixed base resin and curing agent: Experiments and molecular simulation
Okabe, Tomonaga and Takehara, Tomohiro and Inose, Keisuke and Hirano, Noriyuki and Nishikawa, Masaaki and Uehara, Takuya. Curing reaction of epoxy resin composed of mixed base resin and curing agent: Experiments and molecular simulation. Polymer
-
[19]
Chemical reactions in classical molecular dynamics
Gissinger, Jacob R and Jensen, Benjamin D and Wise, Kristopher E. Chemical reactions in classical molecular dynamics. Polymer
-
[20]
Kinetics of the interfacial curing reaction for an epoxy-amine mixture
Yamaguchi, Ko and Kawaguchi, Daisuke and Miyata, Noboru and Miyazaki, Tsukasa and Aoki, Hiroyuki and Yamamoto, Satoru and Tanaka, Keiji. Kinetics of the interfacial curing reaction for an epoxy-amine mixture. Phys. Chem. Chem. Phys
-
[21]
Effect of water content on the thermal degradation of amorphous polyamide 6,6: A collective variable-driven hyperdynamics study
Arash, Behrouz and Thijsse, Barend J and Pecenko, Alessandro and Simone, Angelo. Effect of water content on the thermal degradation of amorphous polyamide 6,6: A collective variable-driven hyperdynamics study. Polym. Degrad. Stab
-
[22]
Resin filling into nano-sized pore on metal surface analyzed by all-atom molecular dynamics simulation over a variety of resin and pore sizes
Mori, Hodaka and Matubayasi, Nobuyuki. Resin filling into nano-sized pore on metal surface analyzed by all-atom molecular dynamics simulation over a variety of resin and pore sizes. Polymer
-
[23]
Molecular dynamics modeling of epoxy resins using the reactive interface force field
Odegard, Gregory M and Patil, Sagar U and Deshpande, Prathamesh P and Kanhaiya, Krishan and Winetrout, Jordan J and Heinz, Hendrik and Shah, Sagar P and Maiaru, Marianna. Molecular dynamics modeling of epoxy resins using the reactive interface force field. Macromolecules
-
[24]
A review on molecularly imprinted polymers preparation by computational simulation-aided methods
Liu, Zhimin and Xu, Zhigang and Wang, Dan and Yang, Yuming and Duan, Y and Ma, Liping and Lin, Tao and Liu, Hongcheng. A review on molecularly imprinted polymers preparation by computational simulation-aided methods. Polymers
-
[25]
Molecular dynamics simulation of polyamide-based materials – A review
Krishna, Sanjay and Sreedhar, I and Patel, Chetan M. Molecular dynamics simulation of polyamide-based materials – A review. Comput. Mater. Sci
-
[26]
A review of advancements in coarse-grained molecular dynamics simulations
Joshi, Soumil Y and Deshmukh, Sanket A. A review of advancements in coarse-grained molecular dynamics simulations. Mol. Simul
-
[27]
Modeling and simulations of polymers: A roadmap
Gartner, III, Thomas E and Jayaraman, Arthi. Modeling and simulations of polymers: A roadmap. Macromolecules
-
[28]
Rate coefficients of free-radical polymerization deduced from pulsed laser experiments
Beuermann, Sabine and Buback, Michael. Rate coefficients of free-radical polymerization deduced from pulsed laser experiments. Prog. Polym. Sci
-
[29]
Cure kinetics of several epoxy–amine systems at ambient and high temperatures
Pramanik, Monoj and Fowler, Eric W and Rawlins, James W. Cure kinetics of several epoxy–amine systems at ambient and high temperatures. J Coat Technol Res
-
[30]
Theoretical catalyst screening of multielement alloy catalysts for ammonia synthesis using machine learning potential and generative artificial intelligence
Hisama, Kaoru and Ishikawa, Atsushi and Aspera, Susan Menez and Koyama, Michihisa. Theoretical catalyst screening of multielement alloy catalysts for ammonia synthesis using machine learning potential and generative artificial intelligence. J. Phys. Chem. C Nanomater. Interfaces
-
[31]
Interconnected lamellar 3D semiconductive PCP for rechargeable aqueous zinc battery cathodes
Lin, Zirui and Otake, Ken-Ichi and Kajiwara, Takashi and Hiraide, Shotaro and Nurhuda, Maryam and Packwood, Daniel and Kadota, Kentaro and Sakamoto, Hirotoshi and Kawaguchi, Shogo and Kubota, Yoshiki and Yao, Ming-Shui and Horike, Satoshi and Sun, Xiaoqi and Kitagawa, Susumu. Interconnected lamellar 3D semiconductive PCP for rechargeable aqueous zinc batt...
-
[32]
Molecular dynamics of liquid-electrode interface by integrating Coulomb interaction into universal neural network potential
Hisama, Kaoru and Valadez Huerta, Gerardo and Koyama, Michihisa. Molecular dynamics of liquid-electrode interface by integrating Coulomb interaction into universal neural network potential. J. Comput. Chem
-
[33]
A neural network potential for the IRMOF series and its application for thermal and mechanical behaviors
Tayfuroglu, Omer and Kocak, Abdulkadir and Zorlu, Y. A neural network potential for the IRMOF series and its application for thermal and mechanical behaviors. Phys. Chem. Chem. Phys
-
[34]
Polymer Science and Technology
Ebewele, Robert O. Polymer Science and Technology
-
[35]
Introduction to Polymers
Young, Robert J and Lovell, Peter A. Introduction to Polymers
-
[36]
Handbook of polymers
Wypych, G. Handbook of polymers
-
[37]
Principles of polymer engineering
McCrum, N G and Buckley, C P and Bucknall, C B. Principles of polymer engineering
-
[38]
Rethinking metadynamics: From bias potentials to probability distributions
Invernizzi, Michele and Parrinello, Michele. Rethinking metadynamics: From bias potentials to probability distributions. J. Phys. Chem. Lett
-
[39]
1999 , publisher=
Polymer handbook , author=. 1999 , publisher=
1999
-
[40]
Merging metadynamics into hyperdynamics: accelerated molecular simulations reaching time scales from microseconds to seconds
Bal, Kristof M and Neyts, Erik C. Merging metadynamics into hyperdynamics: accelerated molecular simulations reaching time scales from microseconds to seconds. J. Chem. Theory Comput
-
[41]
Hyperdynamics: Accelerated molecular dynamics of infrequent events
Voter, Arthur F. Hyperdynamics: Accelerated molecular dynamics of infrequent events. Phys. Rev. Lett
-
[42]
Accelerated molecular dynamics with the bond-boost method
Miron, Radu A and Fichthorn, Kristen A. Accelerated molecular dynamics with the bond-boost method. J. Chem. Phys
-
[43]
Stukowski, Alexander , Title =
-
[44]
VMD : visual molecular dynamics
Humphrey, W and Dalke, A and Schulten, K. VMD : visual molecular dynamics. J. Mol. Graph
-
[45]
2013 , publisher=
Fundamentals of controlled/living radical polymerization , author=. 2013 , publisher=
2013
-
[46]
2004 , publisher=
Principles of polymerization , author=. 2004 , publisher=
2004
-
[47]
Chemical Science , volume=
Intramolecular trapping of spiro radicals to produce unusual cyclization products from usual migration substrates , author=. Chemical Science , volume=. 2023 , publisher=
2023
-
[48]
European polymer journal , volume=
Effect of 1, 1-diphenylethylene on the radical polymerization of di-n-butyl itaconate in benzene , author=. European polymer journal , volume=. 2001 , publisher=
2001
-
[49]
2016 , publisher=
CRC handbook of chemistry and physics , author=. 2016 , publisher=
2016
-
[50]
The Journal of Physical Chemistry A , volume=
Accelerated ReaxFF simulations for describing the reactive cross-linking of polymers , author=. The Journal of Physical Chemistry A , volume=. 2018 , publisher=
2018
-
[51]
The Journal of Physical Chemistry B , volume=
OpenMM 8: molecular dynamics simulation with machine learning potentials , author=. The Journal of Physical Chemistry B , volume=. 2023 , publisher=
2023
-
[52]
The Journal of Physical Chemistry C , volume=
DFT simulation of XPS reveals Cu/epoxy polymer interfacial bonding , author=. The Journal of Physical Chemistry C , volume=. 2019 , publisher=
2019
-
[53]
ACS Applied Polymer Materials , volume=
Molecular events for an epoxy--amine system at a copper interface , author=. ACS Applied Polymer Materials , volume=. 2020 , publisher=
2020
-
[54]
Chemical Physics Letters , volume=
A hybrid MC/MD reaction method with rare event-driving mechanism: Atomistic realization of 2-chlorobutane racemization process in DMF solution , author=. Chemical Physics Letters , volume=. 2013 , publisher=
2013
-
[55]
The Journal of Chemical Physics , volume=
A transformation theory of stochastic evolution in Red Moon methodology to time evolution of chemical reaction process in the full atomistic system , author=. The Journal of Chemical Physics , volume=. 2017 , publisher=
2017
-
[56]
1,1-Diphenylethylene (97\ url =
-
[57]
Matlantis, software as a service style material discovery tool
-
[58]
The Journal of Physical Chemistry C , volume=
Development of a reactive force field for simulations on the catalytic conversion of C/H/O molecules on Cu-metal and Cu-oxide surfaces and application to Cu/CuO-based chemical looping , author=. The Journal of Physical Chemistry C , volume=. 2020 , publisher=
2020
-
[59]
The Journal of Physical Chemistry A , volume=
Computational Insights into Tunable Reversible Network Materials: Accelerated ReaxFF Kinetics of Furan-Maleimide Diels--Alder Reactions for Self-Healing and Recyclability , author=. The Journal of Physical Chemistry A , volume=. 2024 , publisher=
2024
-
[60]
Journal of molecular graphics , volume=
VMD: visual molecular dynamics , author=. Journal of molecular graphics , volume=. 1996 , publisher=
1996
-
[61]
Modelling and simulation in materials science and engineering , volume=
Visualization and analysis of atomistic simulation data with OVITO--the Open Visualization Tool , author=. Modelling and simulation in materials science and engineering , volume=
-
[62]
Bulletin of the Korean Chemical Society , volume=
Universal structure conversion method for organic molecules: from atomic connectivity to three-dimensional geometry , author=. Bulletin of the Korean Chemical Society , volume=. 2015 , publisher=
2015
-
[63]
Journal of Chemical Information and Modeling , volume=
Multimodal Bond Reconstruction toward Generative Molecular Design , author=. Journal of Chemical Information and Modeling , volume=. 2026 , publisher=
2026
-
[66]
Physical chemistry chemical physics , volume=
ReacNetGenerator: an automatic reaction network generator for reactive molecular dynamics simulations , author=. Physical chemistry chemical physics , volume=. 2020 , publisher=
2020
-
[67]
Machine learning meets quantum physics , pages=
Message passing neural networks , author=. Machine learning meets quantum physics , pages=. 2020 , publisher=
2020
-
[68]
Current opinion in structural biology , volume=
Hidden markov models , author=. Current opinion in structural biology , volume=. 1996 , publisher=
1996
-
[69]
IEEE transactions on communications , volume=
List Viterbi decoding algorithms with applications , author=. IEEE transactions on communications , volume=. 1994 , publisher=
1994
-
[70]
mendeleev: A Python resource for properties of chemical elements, ions and isotopes , year =
Mentel,. mendeleev: A Python resource for properties of chemical elements, ions and isotopes , year =
-
[71]
RDKit: Open-Source Cheminformatics Software , url =
Landrum, Greg , biburl =. RDKit: Open-Source Cheminformatics Software , url =
-
[72]
0: the sage small molecule force field , author=
Development and benchmarking of open force field 2.0. 0: the sage small molecule force field , author=. Journal of chemical theory and computation , volume=. 2023 , publisher=
2023
-
[73]
IEEE transactions on pattern analysis and machine intelligence , volume=
Towards understanding convergence and generalization of AdamW , author=. IEEE transactions on pattern analysis and machine intelligence , volume=. 2024 , publisher=
2024
-
[74]
Journal of Cheminformatics , volume=
A rule-based algorithm for automatic bond type perception , author=. Journal of Cheminformatics , volume=. 2012 , publisher=
2012
-
[75]
Christoph Loschen , title =. ChemRxiv , volume =. 2018 , doi =. https://chemrxiv.org/doi/pdf/10.26434/chemrxiv.7403630.v2 , abstract =
-
[76]
Journal of Applied Physics , volume=
Bond order predictions using deep neural networks , author=. Journal of Applied Physics , volume=. 2021 , publisher=
2021
-
[77]
P.; Simm, G
Batatia, I.; Kovacs, D. P.; Simm, G. N. C.; Ortner, C.; Csanyi, G. MACE : Higher Order Equivariant Message Passing Neural Networks for Fast and Accurate Force Fields. Advances in Neural Information Processing Systems. 2022
2022
-
[78]
P.; Musaelian, A.; Simm, G
Batatia, I.; Batzner, S.; Kov \'a cs, D. P.; Musaelian, A.; Simm, G. N. C.; Drautz, R.; Ortner, C.; Kozinsky, B.; Cs \'a nyi, G. The Design Space of E(3)-Equivariant Atom-Centered Interatomic Potentials. 2022
2022
-
[79]
Takamoto, S. et al. Towards universal neural network potential for material discovery applicable to arbitrary combination of 45 elements. Nat. Commun. 2022, 13, 2991
2022
-
[80]
Chen, C.; Ong, S. P. A universal graph deep learning interatomic potential for the periodic table. Nat. Comput. Sci. 2022, 2, 718--728
2022
-
[81]
J.; Ceder, G
Deng, B.; Zhong, P.; Jun, K.; Riebesell, J.; Han, K.; Bartel, C. J.; Ceder, G. CHGNet as a pretrained universal neural network potential for charge-informed atomistic modelling. Nature Machine Intelligence 2023, 5, 1031--1041
2023
-
[82]
Wood, B. M.; Dzamba, M.; Fu, X.; Gao, M.; Shuaibi, M.; Barroso-Luque, L.; Abdelmaqsoud, K.; Gharakhanyan, V.; Kitchin, J. R.; Levine, D. S.; others Uma: A family of universal models for atoms. arXiv preprint arXiv:2506.23971 2025,
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.