Reconstructability of evolutionary intermediates in generative epistatic landscapes
Pith reviewed 2026-06-29 02:08 UTC · model grok-4.3
The pith
Maximum-likelihood intermediates from protein endpoints are often statistically atypical of actual evolutionary paths.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
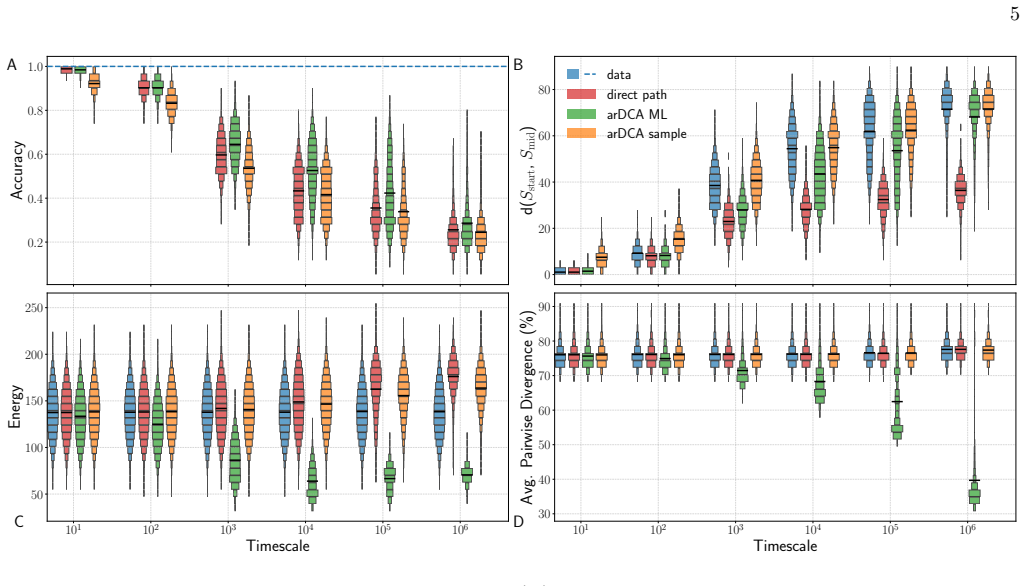
Using generative sequence landscapes as faithful models of protein evolution, maximum-likelihood point predictions for intermediates can match residues yet lie outside the typical distribution of simulated trajectories, whereas conditional sampling recovers the statistical ensemble of histories more faithfully. The amount of recoverable path information depends on the topology of the landscape, with low-mutability regions preserving memory and high-mutability regions opening many alternative routes. Sequence divergence alone is an insufficient clock; incorporating the mutability of the endpoints supplies a more reliable gauge of elapsed evolutionary time. Reconstruction should therefore retu
What carries the argument
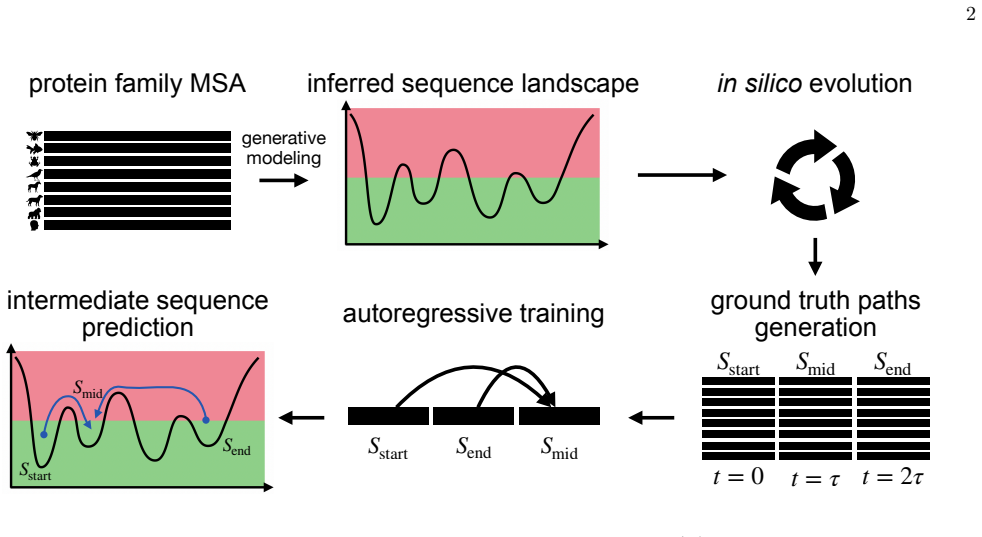
Generative sequence landscapes as controlled models of protein-family evolution that supply ground-truth trajectories for benchmarking reconstruction accuracy.
If this is right
- Maximum-likelihood point estimates should not be treated as typical evolutionary intermediates.
- Conditional sampling from the model yields ensembles that better match the range of plausible histories.
- Low-mutability regions in a landscape preserve more information about the path taken.
- Endpoint mutability must be combined with sequence divergence to estimate elapsed time more accurately.
- Reconstruction methods should return probabilistic ensembles when the landscape topology indicates that multiple histories remain compatible with the endpoints.
Where Pith is reading between the lines
- The same distinction between point estimates and ensembles could be tested on real protein families for which some historical intermediates have been inferred from independent sources.
- If landscape topology controls reconstructability, then experimental evolution in high-mutability versus low-mutability backgrounds should produce measurably different recovery rates.
- The finding that divergence alone misplaces intermediates suggests that existing molecular-clock methods that ignore site-specific mutability may systematically misestimate branch lengths.
- Models trained on one protein family could be used to predict, before data collection, which pairs of extant sequences are likely to retain reconstructible history.
Load-bearing premise
Generative sequence landscapes serve as faithful models of real protein-family evolution so that simulated trajectories can stand in for ground truth.
What would settle it
A direct comparison, on the same protein family, between the distribution of intermediates generated by the model and the distribution recovered from independent phylogenetic or experimental data would show whether the model distributions match real evolutionary statistics.
Figures
read the original abstract
Evolutionary intermediates connect observed proteins, but the sequence of steps that produced them is rarely recoverable from extant data alone. Here we ask what can, and cannot, be inferred about such intermediates from the endpoints. Using generative sequence landscapes as controlled models of protein-family evolution, we benchmark data-driven reconstruction against ground-truth simulated trajectories. We find that the best point prediction is not necessarily the most faithful evolutionary reconstruction: maximum-likelihood intermediates can be residue-wise accurate yet statistically atypical, whereas conditional sampling better captures the ensemble of plausible histories. Predictability is limited by the topology of the landscape. Constrained, low-mutability regions preserve information about the path, while permissive high-mutability regions open many alternative routes and erase path-specific memory. We also show that sequence divergence alone is an insufficient measure of elapsed evolutionary time; incorporating endpoint mutability provides a more reliable way to place intermediates in the landscape. These results recast intermediate reconstruction as a calibrated probabilistic problem. Rather than seeking a single "true" sequence, data-driven models should identify when endpoints contain evolutionary information, and return realistic ensembles.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The paper claims that evolutionary intermediates in protein families cannot be reliably recovered from endpoints alone. Using generative sequence landscapes as controlled models, the authors simulate trajectories as ground truth and benchmark reconstruction methods. They report that maximum-likelihood point estimates can be residue-wise accurate yet statistically atypical of the true ensemble, while conditional sampling better recovers the distribution of plausible histories. Predictability depends on landscape topology: low-mutability constrained regions retain path information, whereas high-mutability permissive regions erase it. Sequence divergence is shown to be an insufficient clock; incorporating endpoint mutability yields a more reliable placement of intermediates. The work concludes that intermediate reconstruction should be treated as a calibrated probabilistic task that returns realistic ensembles rather than single sequences.
Significance. If the simulation-based results hold under the stated experimental design, the paper makes a useful contribution by demonstrating concrete limitations of point-estimate reconstructions and by providing a controlled benchmark for probabilistic alternatives. The explicit use of generative models to generate ground-truth trajectories is a methodological strength that allows direct quantification of reconstruction fidelity without circularity. The findings could inform the design of phylogenetic and ancestral-sequence methods that incorporate ensemble outputs and mutability-aware timing.
major comments (2)
- [Abstract] Abstract: The abstract states clear findings but supplies no quantitative results, model details, error bars, or exclusion criteria, so it is impossible to verify whether the data support the claims as stated. The central claims about residue-wise accuracy versus statistical typicality and the superiority of conditional sampling require at least summary statistics (e.g., mean accuracy, KL divergence between ensembles) to be load-bearing.
- [Methods / Results] Methods / Results (simulation protocol): The benchmarking relies on trajectories simulated inside the same generative landscapes used for reconstruction. While internally consistent, the manuscript should report sensitivity analyses showing that the reported differences between ML and conditional sampling persist across different generative-model hyperparameters or landscape ruggedness levels; otherwise the topology-dependent predictability claim rests on a single model class.
minor comments (3)
- [Abstract] Abstract: Add one or two quantitative effect sizes (e.g., typical accuracy or divergence values) to make the magnitude of the reported phenomena concrete.
- [Abstract / Introduction] Notation: Define 'statistically atypical' and 'endpoint mutability' explicitly on first use; the current phrasing leaves open whether these are residue-frequency deviations or full-sequence likelihood ratios.
- [Figures] Figures: Ensure that any trajectory or ensemble plots include error bars or credible intervals derived from multiple independent simulations.
Simulated Author's Rebuttal
We thank the referee for the positive evaluation and the helpful suggestions for strengthening the manuscript. We address each major comment below and have revised the paper accordingly.
read point-by-point responses
-
Referee: [Abstract] Abstract: The abstract states clear findings but supplies no quantitative results, model details, error bars, or exclusion criteria, so it is impossible to verify whether the data support the claims as stated. The central claims about residue-wise accuracy versus statistical typicality and the superiority of conditional sampling require at least summary statistics (e.g., mean accuracy, KL divergence between ensembles) to be load-bearing.
Authors: We agree that the abstract would be strengthened by quantitative support. In the revised version we have added concise summary statistics (mean residue-wise accuracy with standard error, mean KL divergence between reconstructed and true ensembles, and the fraction of trajectories for which conditional sampling outperforms ML), along with brief model-class and exclusion-criterion statements. These additions remain within the journal’s abstract length limit while making the central claims verifiable from the abstract alone. revision: yes
-
Referee: [Methods / Results] Methods / Results (simulation protocol): The benchmarking relies on trajectories simulated inside the same generative landscapes used for reconstruction. While internally consistent, the manuscript should report sensitivity analyses showing that the reported differences between ML and conditional sampling persist across different generative-model hyperparameters or landscape ruggedness levels; otherwise the topology-dependent predictability claim rests on a single model class.
Authors: The referee correctly identifies that our primary benchmark uses trajectories generated from the same class of models employed for reconstruction. By design this supplies ground-truth paths without circularity, yet we acknowledge the value of broader sensitivity checks. We have therefore added a new supplementary section that repeats the full reconstruction pipeline under (i) varied epistasis strengths, (ii) different mutation-rate regimes, and (iii) two additional ruggedness levels (low and high). The superiority of conditional sampling and the topology dependence remain qualitatively unchanged across these conditions; quantitative differences are reported in new Tables S3–S5. Exhaustive sampling of every conceivable ruggedness parameterization is computationally prohibitive, but the added analyses cover the range of landscape topologies discussed in the main text. revision: partial
Circularity Check
No significant circularity; simulation study is self-contained
full rationale
The paper frames generative sequence landscapes as controlled simulation environments, generates independent trajectories as ground-truth evolutionary histories, and then benchmarks reconstruction methods (ML point estimates vs. conditional sampling) against those trajectories. All central claims—residue accuracy vs. statistical typicality, role of landscape topology and mutability, insufficiency of divergence alone—are direct empirical comparisons within this setup. No derivation reduces a prediction to a fitted quantity by construction, no load-bearing self-citation chain is invoked, and no ansatz or uniqueness theorem is smuggled in. The setup is a standard in silico benchmark whose results hold by design of the experiment rather than by definitional equivalence.
Axiom & Free-Parameter Ledger
Reference graph
Works this paper leans on
-
[1]
Our third main result concerns the transition from sim- ulation to real applications
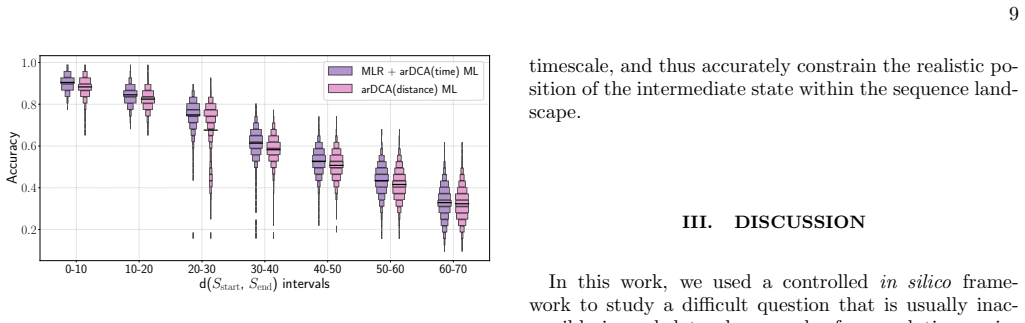
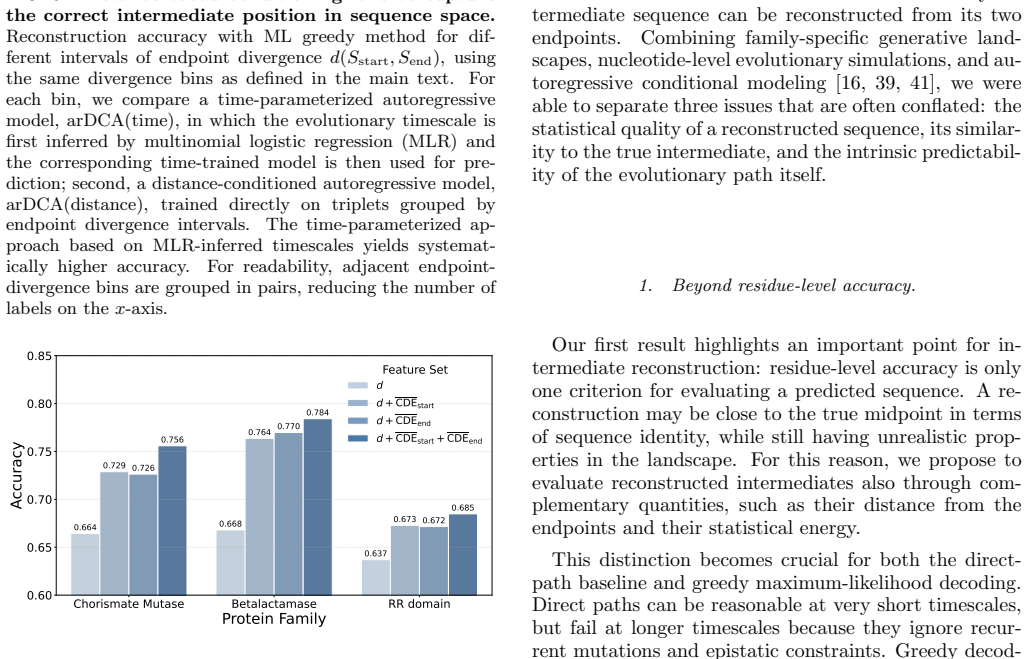
From sequence divergence to evolutionary time. Our third main result concerns the transition from sim- ulation to real applications. In natural data, the elapsed evolutionary time between two sequences is unknown, whereas sequence divergence is directly observable. How- ever, our results show that training reconstruction mod- els based on endpoint diverge...
-
[2]
Taken together, our results provide some interesting answers to the question posed in this work
Conclusion and Future Perspectives. Taken together, our results provide some interesting answers to the question posed in this work. Intermediate evolutionary sequences can be reconstructed in a statisti- cally meaningful way, but only within limited regimes. It is therefore important to ask not only which intermedi- ates are plausible, but also whether t...
-
[3]
Direct path inference As a naive baseline, we define a reconstruction proce- dure based on direct-path interpolation between the two boundary sequences. Intermediate sequences are sam- pled under an independent-site assumption: at each poly- morphic site betweenS start andS end, the two amino acids observed in the endpoints are assigned equal probability ...
-
[4]
The original data are triplets of the form (Sstart, Smid, Send)
Autoregressive modeling As a data-driven reconstruction approach, we use arDCA to learn from the simulated trajectory triplets [41]. The original data are triplets of the form (Sstart, Smid, Send). Since our goal is to predict the in- termediate sequence from the two endpoints, each triplet is reorganized in the order (S start, Send, Smid). With this orde...
-
[5]
Greedy maximum likelihood Once the arDCA model has been trained, different de- coding strategies can be used to reconstruct the evo- lutionary intermediate. The most direct determinis- tic strategy is greedy maximum-likelihood decoding, in which the intermediate sequence is built site by site by se- lecting, at each step, the amino acid with maximum con- ...
-
[6]
Sampling is performed autoregressively, residue by residue, using the same conditional probabilities defined above
Generative sampling Intermediate sequences can also be generated by direct sampling from the conditional distribution learned by the arDCA model, Smid ∼P(S mid|Sstart, Send). Sampling is performed autoregressively, residue by residue, using the same conditional probabilities defined above. In the present work, all samples drawn from the autoregressive mod...
-
[7]
The correspond- ing target distribution becomesP(S mid|Sendpoint), writ- ten as a product of conditional probabilities of the form P(s mid i |S mid <i , Sendpoint)
Model conditioned to one endpoint For the ablation study in which the model is condi- tioned only on the starting point or only on the endpoint, the autoregressive framework is modified by removing one of the two conditioning sequences. The correspond- ing target distribution becomesP(S mid|Sendpoint), writ- ten as a product of conditional probabilities o...
-
[8]
Distance-conditioned training For the distance-conditioned reconstruction analysis, we used the Chorismate Mutase trajectory data and grouped triplets according to the Hamming distance be- tween the two endpoint sequences,d(S start, Send), rather than according to the evolutionary timescaleτ. Training, validation, and test sets were handled separately: wi...
-
[9]
W. P. Russ, M. Figliuzzi, C. Stocker, P. Barrat-Charlaix, M. Socolich, P. Kast, D. Hilvert, R. Monasson, S. Cocco, M. Weigt, and R. Ranganathan, An evolution-based model for designing chorismate mutase enzymes, Science 369, 440 (2020)
2020
-
[10]
McGee, S
F. McGee, S. Hauri, Q. Novinger, S. Vucetic, R. M. Levy, V. Carnevale, and A. Haldane, The generative capacity of probabilistic protein sequence models, Nature Com- munications12, 6302 (2021)
2021
-
[11]
Rehan, E
A. Rehan, E. Mauri, J. Fernandez-de Cossio-Diaz, P.-G. Brun, R. Monasson, M. Ribezzi-Crivellari, and S. Cocco, Design and experimental characterization of specificity- switching mutational paths of WW domains, eLife14, RP110491 (2026), reviewed Preprint
2026
-
[12]
Expanding functional protein sequence space using high entropy generative models
R. Netti, E. Hinds, F. Calvanese, R. Ranganathan, M. Weigt, and F. Zamponi, Expanding functional protein sequence space using high entropy genera- tive models, arXiv 10.48550/arXiv.2605.03578 (2026), arXiv:2605.03578 [q-bio.QM]
work page internal anchor Pith review Pith/arXiv arXiv doi:10.48550/arxiv.2605.03578 2026
-
[13]
Hawkins-Hooker, F
A. Hawkins-Hooker, F. Depardieu, S. Baur, G. Couairon, A. Chen, and D. Bikard, Generating functional protein variants with variational autoencoders, PLOS Computa- tional Biology17, e1008736 (2021)
2021
-
[14]
Madani, B
A. Madani, B. Krause, E. R. Greene, S. Subramanian, B. P. Mohr, J. M. Holton, J. L. Olmos, C. Xiong, Z. Z. Sun, R. Socher, J. S. Fraser, and N. Naik, Large language models generate functional protein sequences across di- verse families, Nature Biotechnology41, 1099 (2023)
2023
-
[15]
Dauparas, I
J. Dauparas, I. Anishchenko, N. Bennett, H. Bai, R. J. Ragotte, L. F. Milles, B. I. M. Wicky, A. Courbet, R. J. de Haas, N. Bethel, P. J. Y. Leung, T. F. Huddy, S. Pel- lock, D. Tischer, F. Chan, B. Koepnick, H. Nguyen, A. Kang, B. Sankaran, A. K. Bera, N. P. King, and D. Baker, Robust deep learning-based protein sequence design using ProteinMPNN, Science...
2022
-
[16]
Nijkamp, J
E. Nijkamp, J. A. Ruffolo, E. N. Weinstein, N. Naik, and A. Madani, Progen2: exploring the boundaries of protein language models, Cell Systems14, 968 (2023)
2023
-
[17]
Ferruz, S
N. Ferruz, S. Schmidt, and B. H¨ ocker, ProtGPT2 is a deep unsupervised language model for protein design, Nature Communications13, 4348 (2022)
2022
-
[18]
F. J. Poelwijk, M. Socolich, and R. Ranganathan, Learn- ing the pattern of epistasis linking genotype and phe- notype in a protein, Nature Communications10, 4213 (2019)
2019
-
[19]
D. M. Weinreich, N. F. Delaney, M. A. DePristo, and D. L. Hartl, Darwinian evolution can follow only very few mutational paths to fitter proteins, Science312, 111 (2006)
2006
-
[20]
Mauri, S
E. Mauri, S. Cocco, and R. Monasson, Mutational paths with sequence-based models of proteins: From sampling to mean-field characterization, Physical Review Letters 130, 158402 (2023)
2023
-
[21]
P. Kantroo, G. P. Wagner, and B. B. Machta, High fitness paths can connect proteins with low sequence overlap, bioRxiv 10.1101/2024.11.13.623265 (2024)
-
[22]
Voordeckers, C
K. Voordeckers, C. A. Brown, K. Vanneste, E. van der Zande, A. Voet, S. Maere, and K. J. Verstrepen, Recon- struction of ancestral metabolic enzymes reveals molec- ular mechanisms underlying evolutionary innovation through gene duplication, PLOS Biology10, e1001446 (2012)
2012
-
[23]
Tian and R
P. Tian and R. B. Best, Exploring the sequence fitness landscape of a bridge between protein folds, PLOS Com- putational Biology16, e1008285 (2020)
2020
-
[24]
Di Bari, M
L. Di Bari, M. Bisardi, S. Cotogno, M. Weigt, and F. Zamponi, Emergent time scales of epistasis in pro- tein evolution, Proceedings of the National Academy of Sciences121, e2406807121 (2024)
2024
-
[25]
J. P. Barton, N. Goonetilleke, T. C. Butler, B. D. Walker, A. J. McMichael, and A. K. Chakraborty, Relative rate and location of intra-host hiv evolution to evade cellu- lar immunity are predictable, Nature Communications 7, 11660 (2016)
2016
-
[26]
Bisardi, J
M. Bisardi, J. Rodriguez-Rivas, F. Zamponi, and M. Weigt, Modeling sequence-space exploration and emergence of epistatic signals in protein evolution, Molec- ular Biology and Evolution39, msab321 (2022)
2022
-
[27]
Biswas, I
A. Biswas, I. Choudhuri, E. Arnold, D. Lyumkis, A. Hal- dane, and R. M. Levy, Kinetic coevolutionary models predict the temporal emergence of hiv-1 resistance mu- tations under drug selection pressure, Proceedings of the National Academy of Sciences121, e2316662121 (2024)
2024
-
[28]
Alvarez, C
S. Alvarez, C. M. Nartey, N. Mercado, J. A. de la Paz, T. Huseinbegovic, and F. Morcos, In vivo functional phe- notypes from a computational epistatic model of evolu- tion, Proceedings of the National Academy of Sciences 121, e2308895121 (2024)
2024
-
[29]
Rossi, L
S. Rossi, L. Di Bari, M. Weigt, and F. Zamponi, Fluctua- tions and the limit of predictability in protein evolution, Reports on Progress in Physics88, 078102 (2025)
2025
-
[30]
J. W. Thornton, Resurrecting ancient genes: experimen- tal analysis of extinct molecules, Nature Reviews Genet- ics5, 366 (2004)
2004
-
[31]
Hofreiter, D
M. Hofreiter, D. Serre, H. N. Poinar, M. Kuch, and S. P¨ a¨ abo, Ancient DNA, Nature Reviews Genetics2, 353 (2001)
2001
-
[32]
P. A. Romero and F. H. Arnold, Exploring protein fit- ness landscapes by directed evolution, Nature Reviews Molecular Cell Biology10, 866 (2009)
2009
-
[33]
Fantini, S
M. Fantini, S. Lisi, P. De Los Rios, A. Cattaneo, and A. Pastore, Protein structural information and evolution- ary landscape by in vitro evolution, Molecular Biology and Evolution37, 1179 (2020)
2020
-
[34]
M. A. Stiffler, F. J. Poelwijk, K. P. Brock, R. R. Stein, A. J. Riesselman, J. Teyra, S. S. Sidhu, D. S. Marks, N. P. Gauthier, and C. Sander, Protein structure from experimental evolution, Cell Systems10, 15 (2020). 15
2020
-
[35]
P. V. Markov, M. Ghafari, M. Beer, K. Lythgoe, P. Sim- monds, N. I. Stilianakis, and A. Katzourakis, The evolu- tion of SARS-CoV-2, Nature Reviews Microbiology21, 361 (2023)
2023
-
[36]
L. Di Bari, T. Mora, A. Pagnani, A. M. Walczak, F. Zam- poni, and S. Rossi, Modeling protein evolution via gen- erative inference from monte carlo chains to population genetics (2026), arXiv:2602.08641 [q-bio.PE]
-
[37]
D. M. Weinreich, R. A. Watson, and L. Chao, Perspec- tive: Sign epistasis and genetic constraint on evolution- ary trajectories, Evolution59, 1165 (2005)
2005
-
[38]
T. N. Starr and J. W. Thornton, Epistasis in protein evolution, Protein Science25, 1204 (2016)
2016
-
[39]
K. Buda, C. M. Miton, and N. Tokuriki, Pervasive epista- sis exposes intramolecular networks in adaptive enzyme evolution, Nature Communications14, 8508 (2023)
2023
-
[40]
Y. Park, B. P. H. Metzger, and J. W. Thornton, Epistatic drift causes gradual decay of predictability in protein evo- lution, Science376, 823 (2022)
2022
-
[41]
Z. Yang, S. Kumar, and M. Nei, A new method of infer- ence of ancestral nucleotide and amino acid sequences., Genetics141, 1641 (1995)
1995
-
[42]
Pupko, I
T. Pupko, I. Pe, R. Shamir, and D. Graur, A fast al- gorithm for joint reconstruction of ancestral amino acid sequences, Molecular Biology and Evolution17, 890 (2000)
2000
-
[43]
Hanson-Smith, B
V. Hanson-Smith, B. Kolaczkowski, and J. W. Thornton, Robustness of ancestral sequence reconstruction to phy- logenetic uncertainty, Molecular Biology and Evolution 27, 1988 (2010)
1988
-
[44]
J. B. Joy, R. H. Liang, R. M. McCloskey, T. Nguyen, and A. F. Poon, Ancestral reconstruction, PLoS Compu- tational Biology12, e1004763 (2016)
2016
-
[45]
Figliuzzi, H
M. Figliuzzi, H. Jacquier, A. Schug, O. Tenaillon, and M. Weigt, Coevolutionary landscape inference and the context-dependence of mutations in beta-lactamase TEM-1, Molecular Biology and Evolution33, 268 (2016)
2016
-
[46]
Morcos, A
F. Morcos, A. Pagnani, B. Lunt, A. Bertolino, D. S. Marks, C. Sander, R. Zecchina, J. N. Onuchic, T. Hwa, and M. Weigt, Direct-coupling analysis of residue coevo- lution captures native contacts across many protein fam- ilies, Proceedings of the National Academy of Sciences 108, E1293 (2011)
2011
-
[47]
Cocco, C
S. Cocco, C. Feinauer, M. Figliuzzi, R. Monasson, and M. Weigt, Inverse statistical physics of protein sequences: A key issues review, Reports on Progress in Physics81, 032601 (2018)
2018
-
[48]
Calvanese, G
F. Calvanese, G. Peinetti, P. Pavlinova, P. Nghe, and M. Weigt, Integrating experimental feed- back improves generative models for biological sequences, Nucleic Acids Research53, gkaf832 (2025), https://academic.oup.com/nar/article- pdf/53/16/gkaf832/64201108/gkaf832.pdf
2025
-
[49]
Trinquier, G
J. Trinquier, G. Uguzzoni, A. Pagnani, F. Zamponi, and M. Weigt, Efficient generative modeling of protein sequences using simple autoregressive models, Nature Communications12, 5800 (2021)
2021
-
[50]
A. P. Muntoni, A. Pagnani, M. Weigt, and F. Zam- poni, adabmDCA: Adaptive boltzmann machine learn- ing for biological sequences, BMC Bioinformatics22, 528 (2021)
2021
-
[51]
Rosset, R
L. Rosset, R. Netti, A. P. Muntoni, M. Weigt, and F. Zamponi, adabmDCA 2.0—a flexible but easy-to-use package for direct coupling analysis, inProtein Evolution: Methods and Protocols, Methods in Molecular Biology, Vol. 2979, edited by S. M. Khan and F. Pazos (Humana, New York, NY, 2026) pp. 83–104
2026
-
[52]
P. D. Williams, D. D. Pollock, B. P. Blackburne, and R. A. Goldstein, Assessing the accuracy of ancestral pro- tein reconstruction methods, PLOS Computational Biol- ogy2, e69 (2006)
2006
-
[53]
De Leonardis, A
M. De Leonardis, A. Pagnani, and P. Barrat-Charlaix, Reconstruction of ancestral protein sequences using au- toregressive generative models, Molecular Biology and Evolution42, msaf070 (2025)
2025
-
[54]
R. M. Levy, A. Haldane, and W. F. Flynn, Potts hamil- tonian models of protein co-variation, free energy land- scapes, and evolutionary fitness, Current Opinion in Structural Biology43, 55 (2017)
2017
-
[55]
J. A. G. M. de Visser and J. Krug, Empirical fitness land- scapes and the predictability of evolution, Nature Re- views Genetics15, 480 (2014)
2014
-
[56]
Vigu´ e, G
L. Vigu´ e, G. Croce, M. Petitjean, E. Rupp´ e, O. Tenaillon, and M. Weigt, Deciphering polymorphism in 61,157 Es- cherichia coli genomes via epistatic sequence landscapes, Nature Communications13, 4030 (2022)
2022
-
[57]
A. Koehl, S. Prillo, M. Liu, J. Xiong, L. Weng, D. F. Savage, and Y. S. Song, Deep models of protein evolution in time generate realistic evolutionary trajectories and functional proteins, bioRxiv 10.64898/2026.02.19.706898 (2026)
-
[58]
F. J. Poelwijk, V. Krishna, and R. Ranganathan, The context-dependence of mutations: A linkage of for- malisms, PLOS Computational Biology12, e1004771 (2016)
2016
-
[59]
Rodriguez-Rivas, G
J. Rodriguez-Rivas, G. Croce, M. Muscat, and M. Weigt, Epistatic models predict mutable sites in SARS-CoV- 2 proteins and epitopes, Proceedings of the National Academy of Sciences119, e2113118119 (2022)
2022
-
[60]
N. Goldman and Z. Yang, A codon-based model of nu- cleotide substitution for protein-coding dna sequences, Molecular Biology and Evolution11, 725 (1994). 16 SUPPLEMENTARY APPENDIX S1. PROTEIN FAMILIES AND MULTIPLE SEQUENCE ALIGNMENTS S1.1. Chorismate Mutase Chorismate Mutases (CM; Pfam accession: PF01817) are enzymes that catalyze the conversion of choris...
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.