A Transferable Learned Temporal Prior for Transmission Reconstruction and Decision-Relevant Uncertainty in Real Outbreak Labels
Pith reviewed 2026-07-01 06:22 UTC · model grok-4.3
The pith
A temporal prior learned on eleven disease families and locked before seeing target data improves Andes virus parent ranking over baselines.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
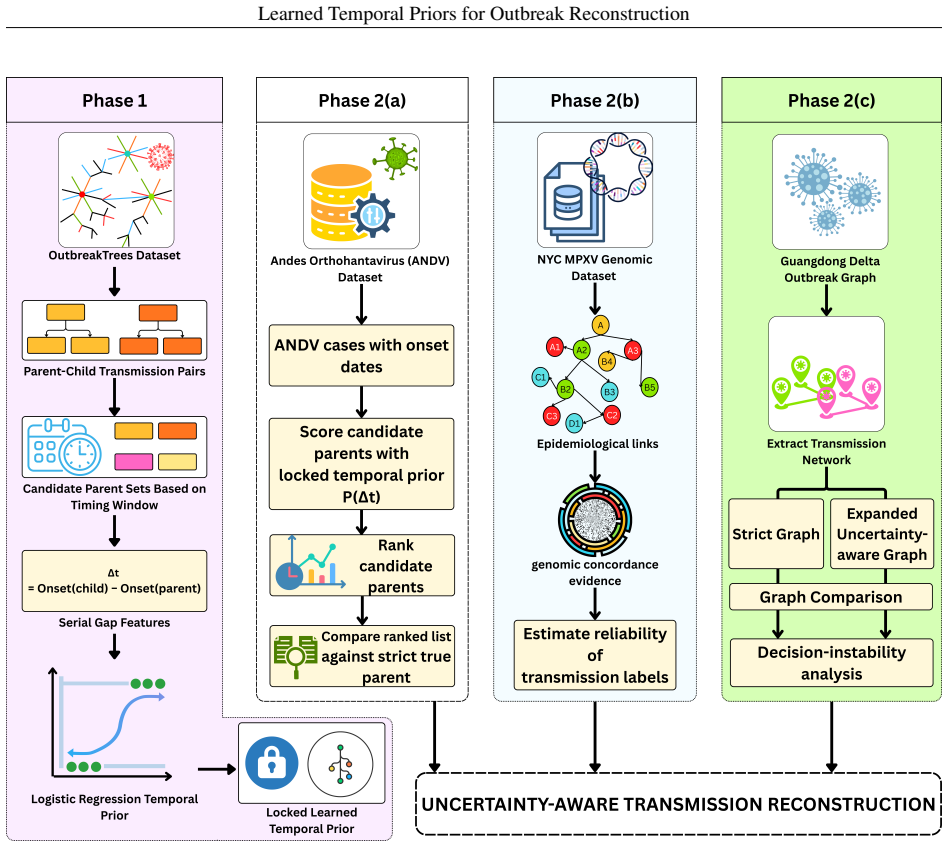
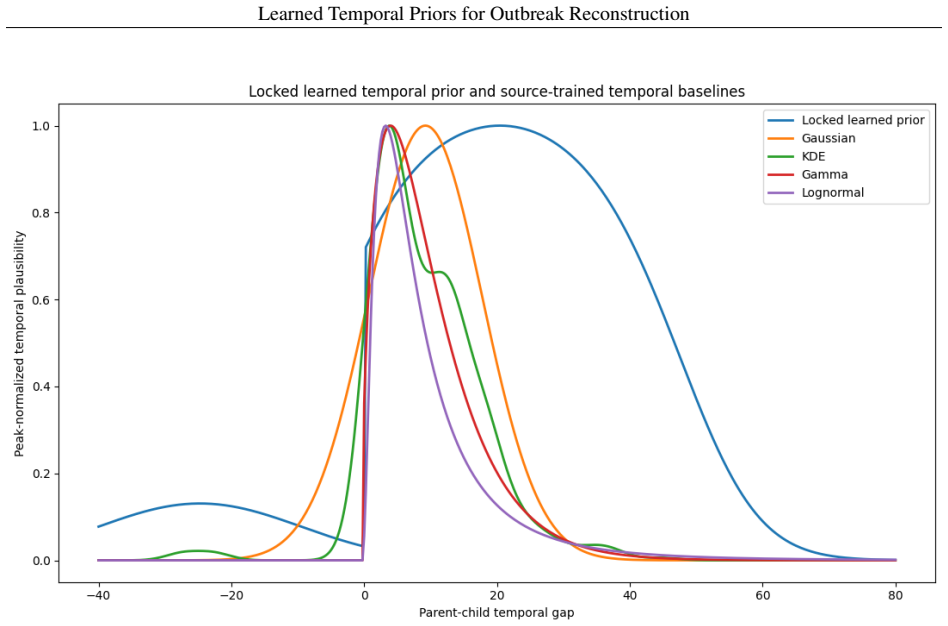
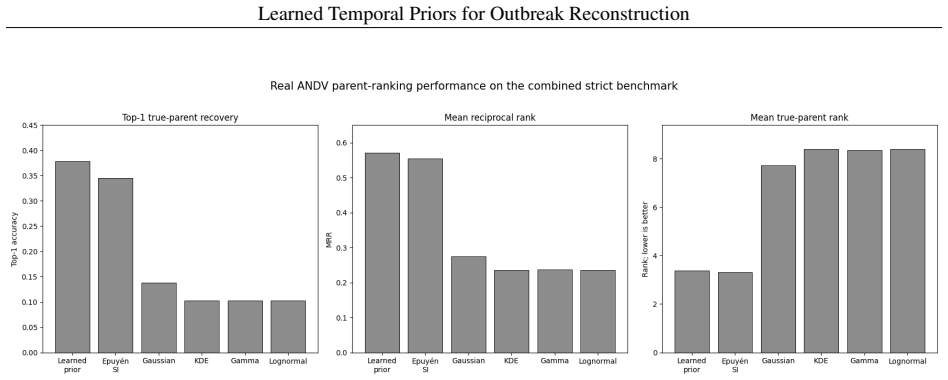
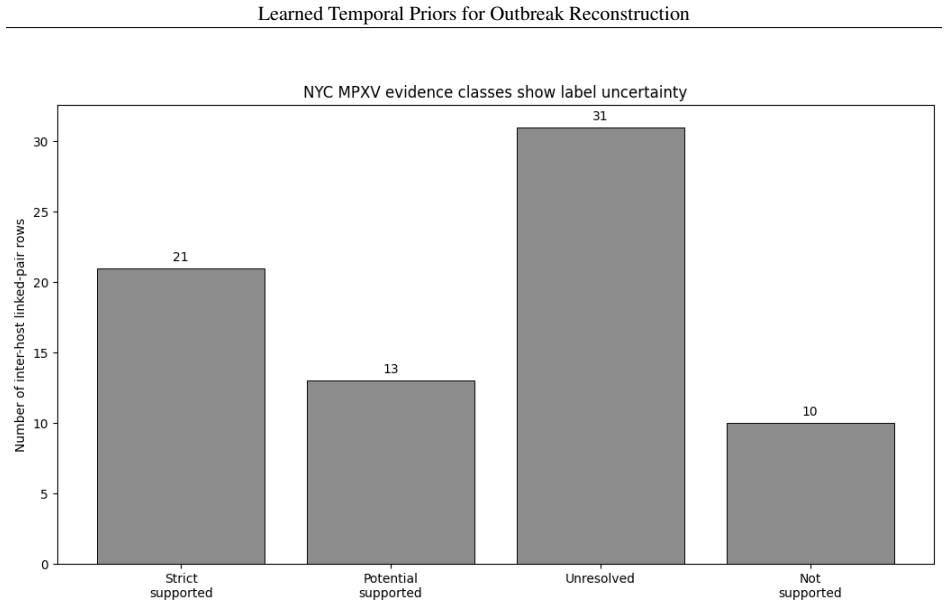
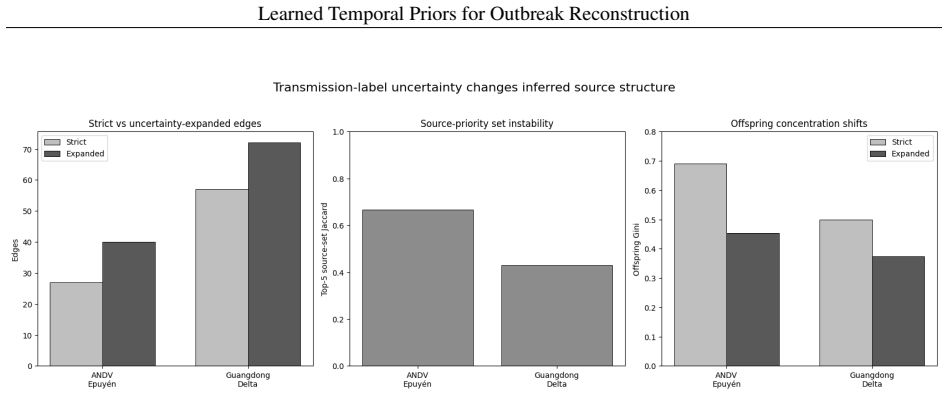
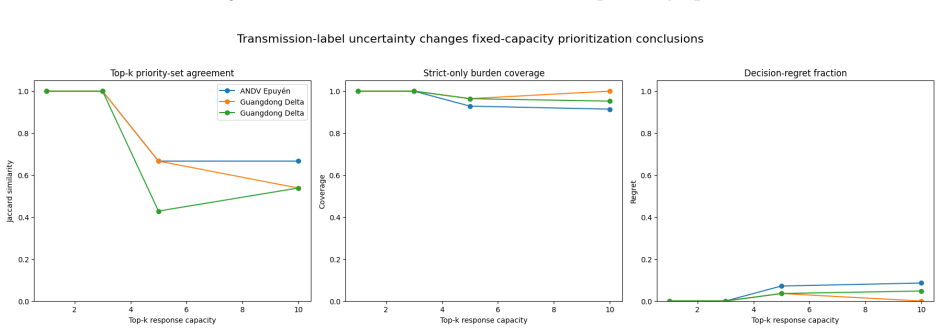
A logistic regression temporal prior trained on eleven disease families, with all parameters locked before any Andes virus data is seen, produces mean reciprocal rank 0.571 and top-1 accuracy 37.9 percent on a 29-task parent-ranking benchmark, compared with 0.274 and 13.8 percent for the best source-trained baseline; permutation tests give p less than or equal to 0.0002. An independent phylogenetic audit of 75 New York City mpox pairs finds 54.67 percent genomically unresolved or unsupported. Retaining those uncertain edges in Andes virus and Guangdong Delta graphs changes the Jaccard overlap of top-5 source-priority sets to between 0.429 and 0.667.
What carries the argument
Locked logistic regression temporal prior trained on eleven disease families and applied without refitting
If this is right
- Source-priority sets for intervention shift when uncertain transmission edges are retained rather than discarded.
- Transmission-label uncertainty can be measured directly from phylogenetic concordance in real outbreak graphs.
- A single locked model can be applied across outbreaks without collecting target-specific training data.
- Current practice of treating epidemiological timing and labels as deterministic ground truth understates reconstruction error.
Where Pith is reading between the lines
- The same locked prior could be tested on additional virus families to check whether the transfer advantage holds beyond Andes virus.
- Decision pipelines that currently drop uncertain edges might instead propagate them as weighted inputs to intervention ranking.
- If phylogenetic audits become routine, outbreak graphs could carry explicit uncertainty flags that change which cases receive follow-up resources.
Load-bearing premise
Temporal transmission patterns learned from the eleven disease families are similar enough to Andes virus dynamics that the frozen model transfers without any adjustment.
What would settle it
A new independent outbreak dataset on which the locked prior produces lower mean reciprocal rank than a source-trained parametric baseline would falsify the transfer claim.
Figures
read the original abstract
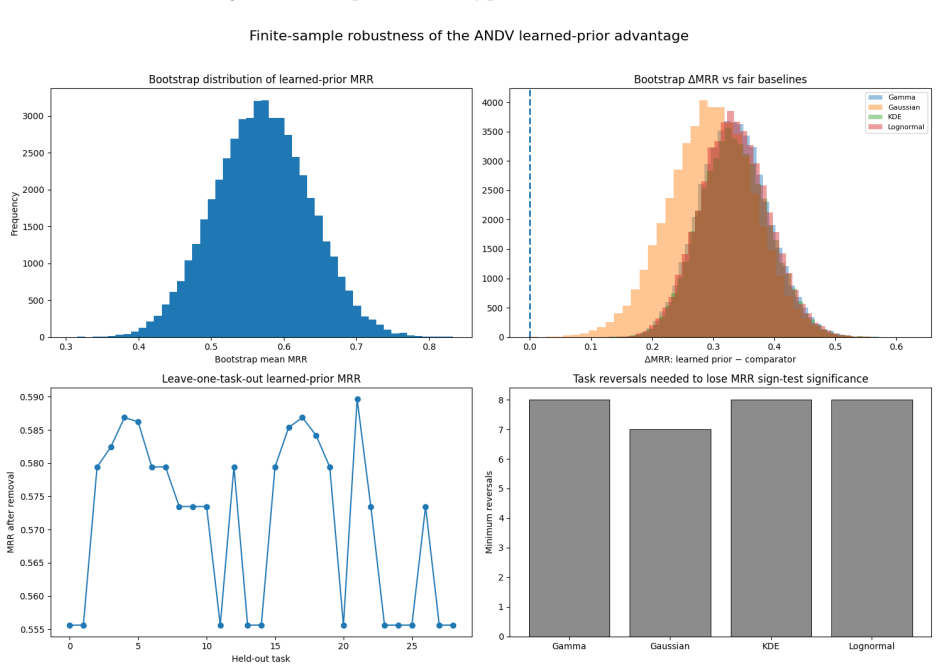
Outbreak transmission reconstruction treats epidemiological timing and transmission labels as deterministic ground truth; neither has been systematically evaluated. We trained a logistic regression temporal prior on eleven disease families, locked all parameters before accessing any target outbreak data, and applied it without refitting to a strict Andes virus (ANDV) parent-ranking benchmark of 29 tasks. The locked prior achieved mean reciprocal rank (MRR) 0.571 versus 0.274 and Top-1 accuracy 37.9% versus 13.8% against the best source-trained parametric baseline (permutation p <= 0.0002; 7-8 reversals to lose MRR significance). A phylogenetic concordance audit of 75 NYC mpox inter-host pairs - independent label-reliability evidence rather than a prior validation - found that 54.67% (exact 95% CI: 42.75-66.21%) were genomically unresolved or unsupported. Retaining uncertain edges in ANDV and Guangdong Delta graphs shifted top-5 source-priority sets (Jaccard 0.429-0.667). Transmission-label uncertainty was measurable in the outbreak evidence modules examined, and retaining uncertain links changed which source cases were prioritized for intervention.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript claims that a logistic regression temporal prior trained on eleven disease families, with all parameters locked before accessing any target data, transfers without refitting to a strict Andes virus (ANDV) parent-ranking benchmark of 29 tasks. It reports improved mean reciprocal rank (0.571 vs. 0.274) and Top-1 accuracy (37.9% vs. 13.8%) over the best source-trained parametric baseline, supported by a permutation test (p ≤ 0.0002). A separate phylogenetic audit of 75 NYC mpox inter-host pairs is presented as evidence of label uncertainty, showing that retaining uncertain edges alters top-5 source-priority sets (Jaccard 0.429-0.667).
Significance. If the transferability claim holds after addressing domain-similarity concerns, the work would offer a practical, locked prior for transmission reconstruction that demonstrably improves ranking performance on real outbreak data while quantifying how label uncertainty affects intervention prioritization. The independent mpox audit and permutation-based significance testing are strengths that support falsifiability of the performance claims.
major comments (1)
- [Abstract and ANDV benchmark description] The central transfer result (MRR 0.571 / Top-1 37.9% on the 29 ANDV tasks) rests on the assumption that temporal patterns learned from the eleven source families align with ANDV dynamics sufficiently for locked application. No comparison of generation-interval distributions, incubation-period statistics, or fitted coefficient stability between source families and ANDV is described in the benchmark or methods sections; the permutation test only contrasts against the reported baseline and does not test whether the baseline would improve under ANDV-specific temporal statistics. This assumption is load-bearing for the claim of a generalizable transferable prior.
minor comments (2)
- [Results on phylogenetic audit] The mpox phylogenetic audit is explicitly independent of prior validation; the text should clarify whether any of its uncertainty estimates were used to adjust the ANDV graphs or remain strictly separate.
- [Methods] Feature construction, exact timing variables, and exclusion rules for the eleven source families and the ANDV benchmark are referenced but not detailed enough for full reproducibility of the locked model.
Simulated Author's Rebuttal
We thank the referee for the constructive review and for identifying the load-bearing assumption in the transferability claim. We respond to the single major comment below and indicate planned revisions.
read point-by-point responses
-
Referee: [Abstract and ANDV benchmark description] The central transfer result (MRR 0.571 / Top-1 37.9% on the 29 ANDV tasks) rests on the assumption that temporal patterns learned from the eleven source families align with ANDV dynamics sufficiently for locked application. No comparison of generation-interval distributions, incubation-period statistics, or fitted coefficient stability between source families and ANDV is described in the benchmark or methods sections; the permutation test only contrasts against the reported baseline and does not test whether the baseline would improve under ANDV-specific temporal statistics. This assumption is load-bearing for the claim of a generalizable transferable prior.
Authors: We agree that the manuscript does not include explicit comparisons of generation-interval distributions, incubation-period statistics, or fitted coefficient stability between the eleven source families and ANDV; this omission weakens the supporting evidence for alignment. We will add such comparisons (using available timing metadata from the source families and the ANDV benchmark) to the methods and results sections in revision. At the same time, the primary support for transferability remains the locked-prior performance on the 29 held-out ANDV tasks, which yields a statistically significant gain over the best source-trained parametric baseline under a permutation test that respects the no-target-data constraint. An ANDV-specific version of the baseline would, by definition, require target data and therefore falls outside the locked-prior experimental setting the paper evaluates. The breadth of the eleven source families and the magnitude of the observed improvement (MRR 0.571 vs. 0.274) provide indirect evidence that the learned coefficients capture transferable temporal structure rather than family-specific idiosyncrasies. revision: partial
Circularity Check
No significant circularity detected
full rationale
The paper trains a logistic regression temporal prior on eleven source disease families, explicitly locks all parameters before accessing any ANDV target data, and evaluates transfer performance on an independent 29-task parent-ranking benchmark. The reported MRR and Top-1 metrics are computed on held-out target data rather than being fitted or redefined from the source inputs. The phylogenetic concordance audit on NYC mpox pairs is presented separately as label-reliability evidence and does not enter the prior training or ANDV evaluation equations. No self-definitional reduction, fitted-input prediction, or load-bearing self-citation chain appears in the derivation.
Axiom & Free-Parameter Ledger
free parameters (1)
- logistic regression coefficients
axioms (1)
- domain assumption Temporal patterns of transmission are sufficiently conserved across the eleven disease families to justify transfer to Andes virus without refitting.
Reference graph
Works this paper leans on
-
[1]
Saymon Akther, Michelle Su, Jade C. Wang, Helly Amin, Faten Taki, Nelson De La Cruz, Moinuddin Chowdhury, Tyler Clabby, Erik Kopping, Victoria E. Ruiz, Mindy Leelawong, Julia Latash, Kimberly Johnson, Jennifer Baumgartner, Marcia Wong, Aaron Olsen, Randal C. Fowler, Jonathan E. Pekar, Jennifer L. Havens, Tetyana I. Vasylyeva, Joel O. Wertheim, Scott Hughe...
-
[2]
outbreaker2: a modular platform for outbreak reconstruction.BMC bioinformatics, 19(Suppl 11):363, 2018
Finlay Campbell, Xavier Didelot, Rich Fitzjohn, Neil Ferguson, Anne Cori, and Thibaut Jombart. outbreaker2: a modular platform for outbreak reconstruction.BMC bioinformatics, 19(Suppl 11):363, 2018
2018
-
[3]
Incorporating epidemiological data into the genomic analysis of partially sampled infectious disease outbreaks.Molecular Biology and Evolution, 42(4):msaf083,
Jake Carson, Matt Keeling, Paolo Ribeca, and Xavier Didelot. Incorporating epidemiological data into the genomic analysis of partially sampled infectious disease outbreaks.Molecular Biology and Evolution, 42(4):msaf083,
-
[4]
doi:10.1093/molbev/msaf083
-
[5]
Caroline Colijn, Matthew Hall, and Remco Bouckaert. Taking a BREATH (Bayesian reconstruction and evolu- tionary analysis of transmission histories) to simultaneously infer phylogenetic and transmission trees for partially sampled outbreaks.bioRxiv, July 2024. doi:10.1101/2024.07.11.603095. URL https://doi.org/10.1101/ 2024.07.11.603095. Preprint
-
[6]
Ferguson, Christophe Fraser, and Simon Cauchemez
Anne Cori, Neil M. Ferguson, Christophe Fraser, and Simon Cauchemez. A new framework and software to estimate time-varying reproduction numbers during epidemics.American Journal of Epidemiology, 178(9): 1505–1512, 2013. doi:10.1093/aje/kwt133
-
[7]
Scotti: efficient reconstruction of transmission within outbreaks with the structured coalescent.PLoS computational biology, 12(9):e1005130, 2016
Nicola De Maio, Chieh-Hsi Wu, and Daniel J Wilson. Scotti: efficient reconstruction of transmission within outbreaks with the structured coalescent.PLoS computational biology, 12(9):e1005130, 2016
2016
-
[8]
Bayesian reconstruction of transmission within outbreaks using genomic variants.PLoS computational biology, 14(4):e1006117, 2018
Nicola De Maio, Colin J Worby, Daniel J Wilson, and Nicole Stoesser. Bayesian reconstruction of transmission within outbreaks using genomic variants.PLoS computational biology, 14(4):e1006117, 2018
2018
-
[9]
Bayesian inference of infectious disease transmission from whole-genome sequence data.Molecular biology and evolution, 31(7):1869–1879, 2014
Xavier Didelot, Jennifer Gardy, and Caroline Colijn. Bayesian inference of infectious disease transmission from whole-genome sequence data.Molecular biology and evolution, 31(7):1869–1879, 2014
2014
-
[10]
Genomic infectious disease epidemiology in partially sampled and ongoing outbreaks.Molecular biology and evolution, 34(4):997–1007, 2017
Xavier Didelot, Christophe Fraser, Jennifer Gardy, and Caroline Colijn. Genomic infectious disease epidemiology in partially sampled and ongoing outbreaks.Molecular biology and evolution, 34(4):997–1007, 2017
2017
-
[11]
Estimating the generation interval for coronavirus disease (covid-19) based on symptom onset data, march 2020
Tapiwa Ganyani, Cécile Kremer, Dongxuan Chen, Andrea Torneri, Christel Faes, Jacco Wallinga, and Niel Hens. Estimating the generation interval for coronavirus disease (covid-19) based on symptom onset data, march 2020. Eurosurveillance, 25(17):2000257, 2020
2020
-
[12]
Decision making under deep uncertainty for pandemic policy planning.Health policy, 133:104831, 2023
Sophie Hadjisotiriou, Vincent Marchau, Warren Walker, and Marcel Olde Rikkert. Decision making under deep uncertainty for pandemic policy planning.Health policy, 133:104831, 2023
2023
-
[13]
Zainah Kabami, Alex R. Ario, Julie R. Harris, Mackline Ninsiima, Sherry R. Ahirirwe, Jane R. Aceng Ocero, Diana Atwine, Henry G. Mwebesa, Daniel J. Kyabayinze, Allan N. Muruta, et al. Ebola disease outbreak caused by the sudan virus in uganda, 2022: A descriptive epidemiological study.The Lancet Global Health, 12(10): e1684–e1692, 2024. doi:10.1016/S2214-...
-
[14]
Molecular infectious disease epidemiology: survival analysis and algorithms linking phylogenies to transmission trees.PLoS computational biology, 12(4): e1004869, 2016
Eben Kenah, Tom Britton, M Elizabeth Halloran, and Ira M Longini Jr. Molecular infectious disease epidemiology: survival analysis and algorithms linking phylogenies to transmission trees.PLoS computational biology, 12(4): e1004869, 2016
2016
-
[15]
Aaron A King, Matthieu Domenech de Cellès, Felicia MG Magpantay, and Pejman Rohani. Avoidable errors in the modelling of outbreaks of emerging pathogens, with special reference to ebola.Proceedings of the Royal Society B: Biological Sciences, 282(1806), 2015. doi:https://doi.org/10.1098/rspb.2015.0347
-
[16]
Allan Komakech, Shannon L. M. Whitmer, Jonathan Izudi, Charles Kizito, Mackline Ninsiima, Sherry R. Ahirirwe, Zainah Kabami, Alex R. Ario, et al. Sudan virus disease super-spreading, uganda, 2022.BMC Infectious Diseases, 24:520, 2024. doi:10.1186/s12879-024-09391-0
-
[17]
Viral infection and transmission in a large, well-traced outbreak caused by the sars-cov-2 delta variant.Nature communications, 13(1):460, 2022
Baisheng Li, Aiping Deng, Kuibiao Li, Yao Hu, Zhencui Li, Yaling Shi, Qianling Xiong, Zhe Liu, Qianfang Guo, Lirong Zou, et al. Viral infection and transmission in a large, well-traced outbreak caused by the sars-cov-2 delta variant.Nature communications, 13(1):460, 2022
2022
-
[18]
super-spreaders
Valeria P Martínez, Nicholas Di Paola, Daniel O Alonso, Unai Pérez-Sautu, Carla M Bellomo, Ayelén A Iglesias, Rocio M Coelho, Beatriz López, Natalia Periolo, Peter A Larson, et al. “super-spreaders” and person-to-person transmission of andes virus in argentina.New England Journal of Medicine, 383(23):2230–2241, 2020
2020
-
[19]
Birgit Nikolay, Henrik Salje, M. Jahangir Hossain, A.K.M. Dawlat Khan, Hossain M.S. Sazzad, Mahmudur Rahman, Peter Daszak, Emily S. Gurley, et al. Transmission of nipah virus — 14 years of investigations in bangladesh.New England Journal of Medicine, 380(19):1804–1814, 5 2019. doi:10.1056/NEJMoa1805376. URL https://doi.org/10.1056/NEJMoa1805376
-
[20]
Moreno, Taylor Brock-Fisher, Lydia A
Ivan Specht, Gage K. Moreno, Taylor Brock-Fisher, Lydia A. Krasilnikova, Brittany A. Petros, Jonathan E. Pekar, Mark Schifferli, Ben Fry, Catherine M. Brown, Lawrence C. Madoff, Meagan Burns, Stephen F. Schaffner, Daniel J. Park, Bronwyn L. MacInnis, Al Ozonoff, Patrick Varilly, Michael D. Mitzenmacher, and Pardis C. Sabeti. JUNIPER: Reconstructing transm...
-
[21]
Juliana C. Taube, Paige B. Miller, and John M. Drake. An open-access database of infectious dis- ease transmission trees to explore superspreader epidemiology.PLOS Biology, 20(6):e3001685, 2022. doi:10.1371/journal.pbio.3001685. URLhttps://doi.org/10.1371/journal.pbio.3001685
-
[22]
Vickers and Elena B
Andrew J. Vickers and Elena B. Elkin. Decision curve analysis: A novel method for evaluating prediction models. Medical Decision Making, 26(6):565–574, 2006
2006
-
[23]
Hannah Waddel, Katia Koelle, and Max S. Y . Lau. ScITree: Scalable bayesian inference of transmis- sion tree from epidemiological and genomic data.PLOS Computational Biology, 21(6):e1012657, 2025. doi:10.1371/journal.pcbi.1012657. URLhttps://doi.org/10.1371/journal.pcbi.1012657
-
[24]
How generation intervals shape the relationship between growth rates and reproductive numbers.Proceedings of the Royal Society B: Biological Sciences, 274(1609):599–604, 2007
Jacco Wallinga and Marc Lipsitch. How generation intervals shape the relationship between growth rates and reproductive numbers.Proceedings of the Royal Society B: Biological Sciences, 274(1609):599–604, 2007
2007
-
[25]
Jacco Wallinga and Peter Teunis. Different epidemic curves for severe acute respiratory syndrome reveal similar impacts of control measures.American Journal of Epidemiology, 160(6):509–516, 2004. doi:10.1093/aje/kwh255
-
[26]
Michael Walsh, S. K. Srinathan, Daniel F. McAuley, Marko Mrkobrada, Oron Levine, Christine Ribic, et al. The statistical significance of randomized controlled trial results is frequently fragile: A case for a fragility index. Journal of Clinical Epidemiology, 67(6):622–628, 2014
2014
-
[27]
Wasserstein and Nicole A
Ronald L. Wasserstein and Nicole A. Lazar. The asa’s statement on p-values: Context, process, and purpose.The American Statistician, 70(2):129–133, 2016. 30
2016
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.