Approximating Hartree-Fock theory via an efficiently local reformulation
Pith reviewed 2026-06-28 11:55 UTC · model grok-4.3
The pith
Reorganizing the Hartree-Fock equations pairs each local orbital degree of freedom with a matching local solution condition so that any sparsity pattern can be chosen without losing fast self-consistent field optimization.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
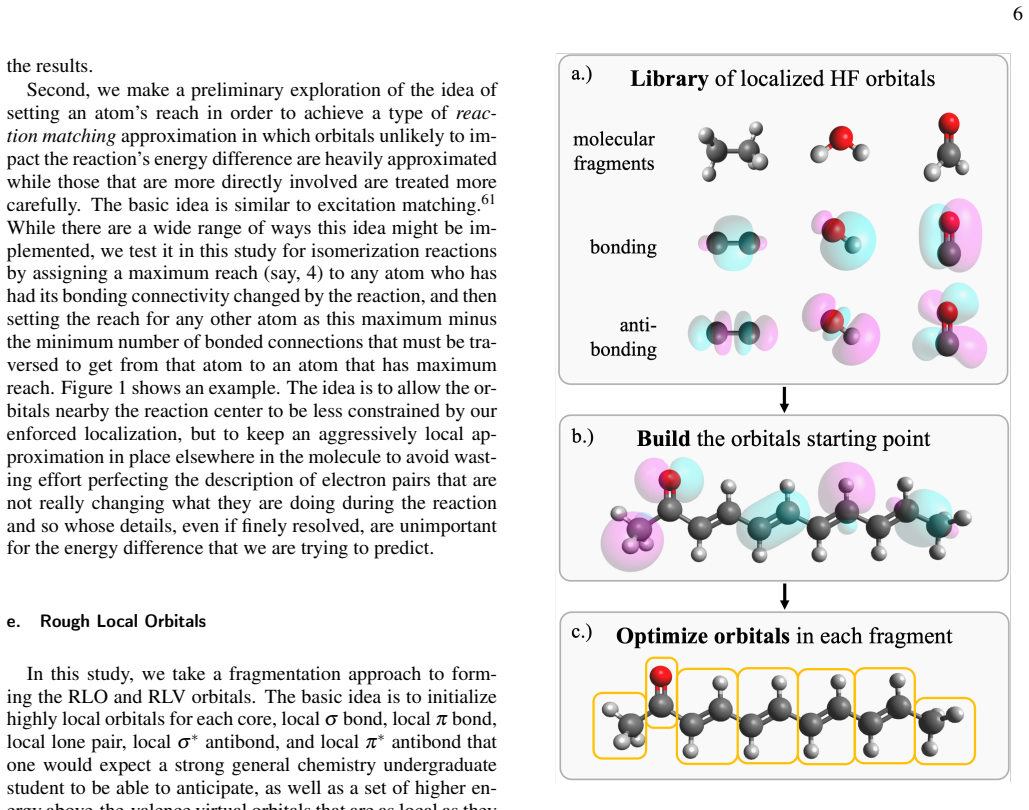
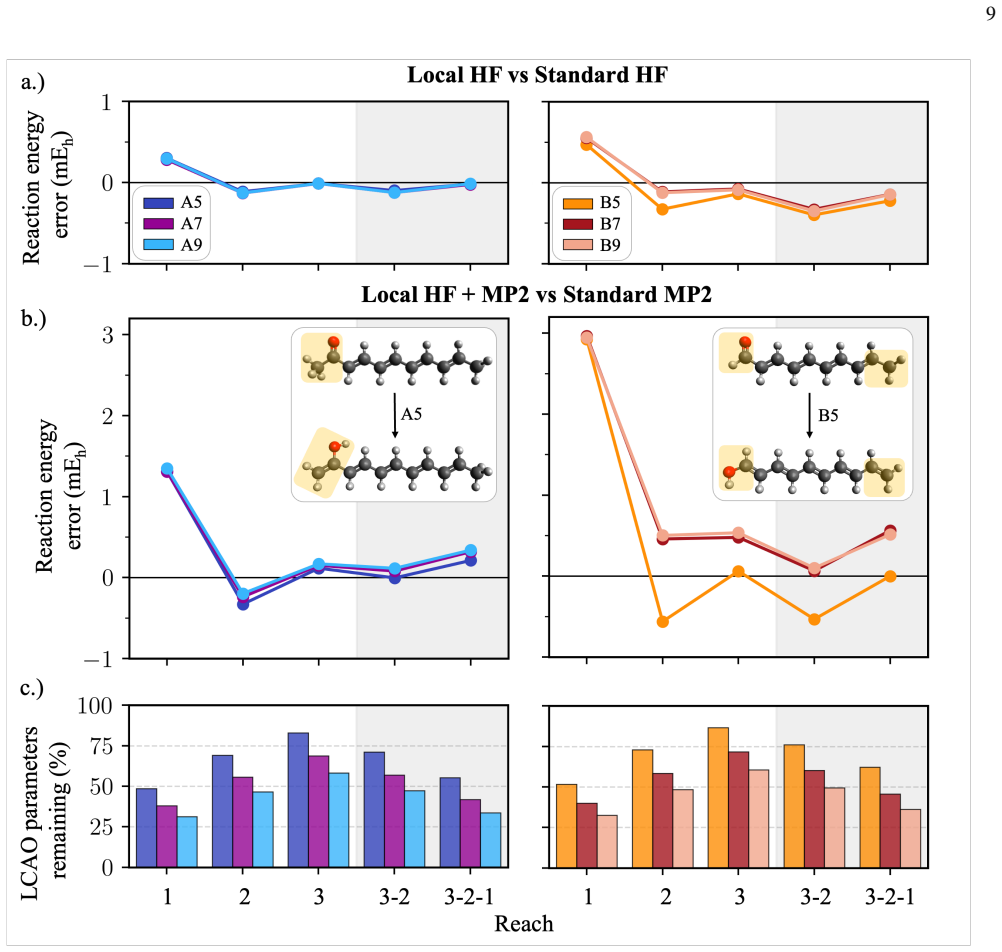
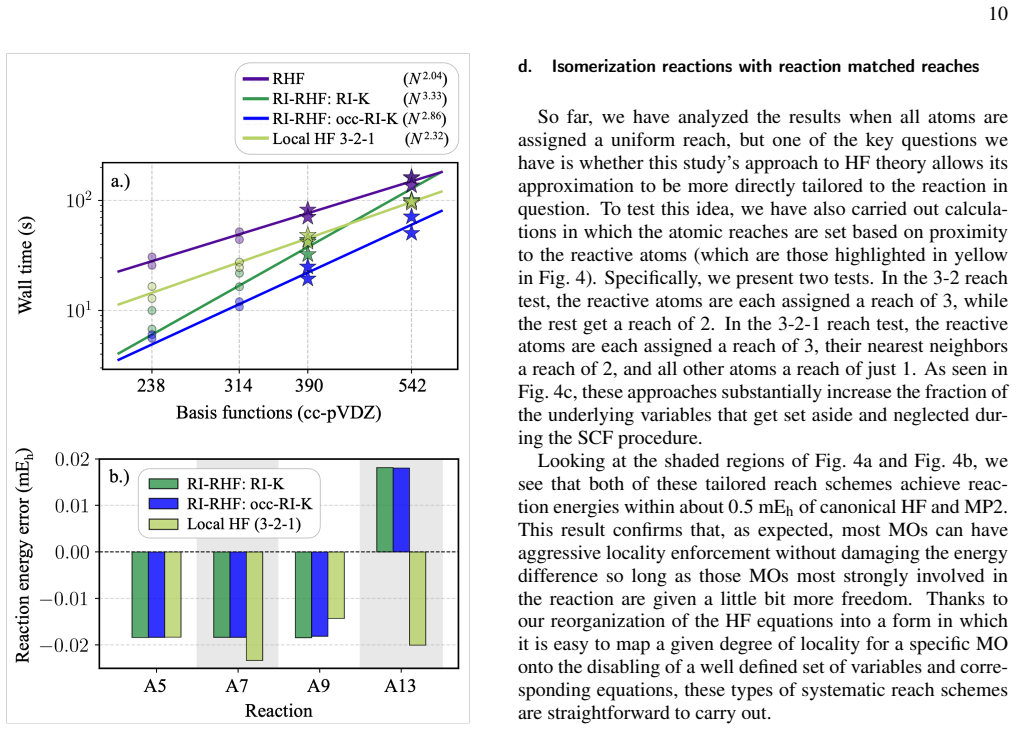
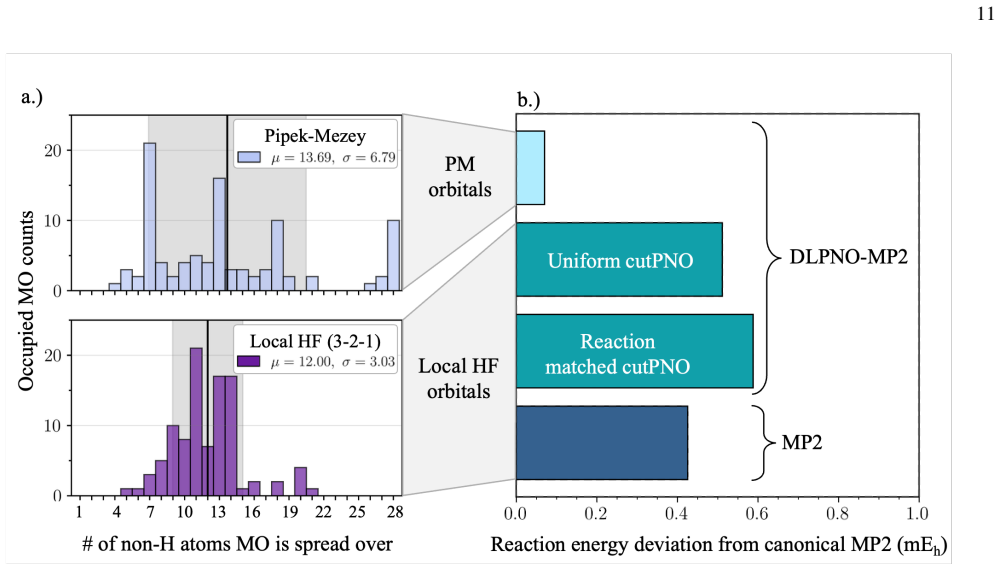
By reorganizing the Hartree-Fock equations, each local degree of freedom in the molecular orbitals is paired with a specific solution condition that has a naturally local interpretation. These pairs can be independently turned on or off according to any chosen sparsity pattern, and the overall self-consistent field optimization remains fast and efficient. This structure is used to impose reaction-matched locality schemes, leading to Fock builds that exploit locality for competitive timings and minimal impact on reaction energy accuracy in Hartree-Fock and MP2 calculations.
What carries the argument
The reorganized pairing of each local orbital degree of freedom with a corresponding local solution condition, which permits independent activation of locality constraints while preserving SCF efficiency.
If this is right
- Any sparsity pattern for orbital locality can be selected without changing the speed of the self-consistent field optimization.
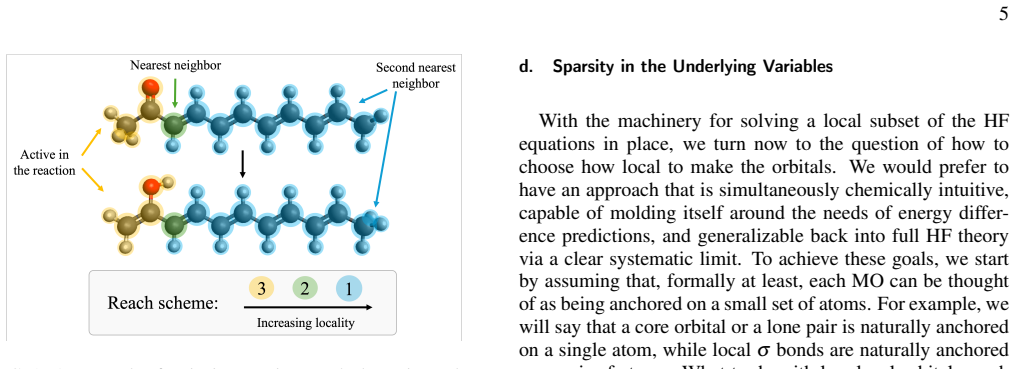
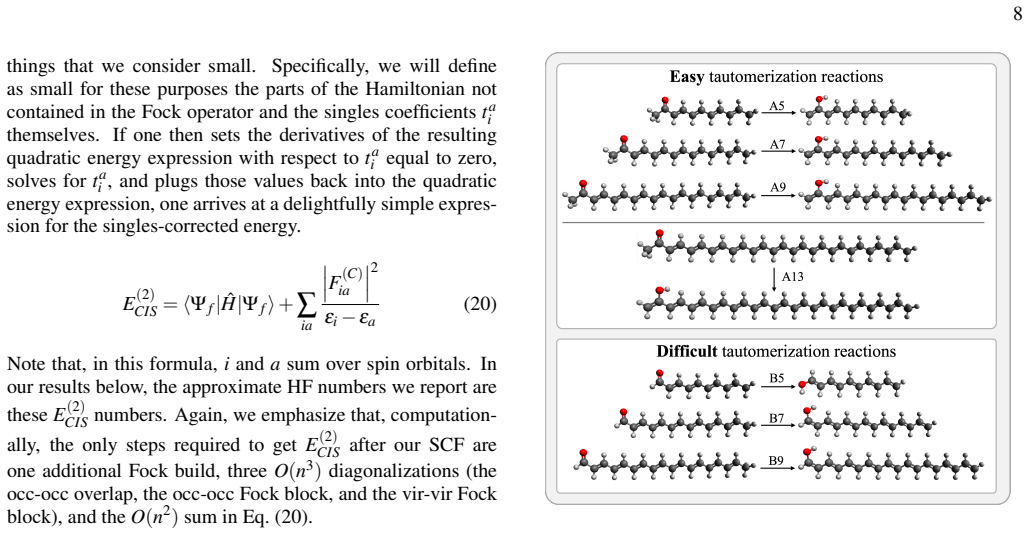
- Reaction-matched locality schemes enable Fock builds that exploit locality to achieve competitive timings in modestly sized molecules.
- Imposed orbital locality arranged in this way causes minimal damage to Hartree-Fock and MP2 reaction energy predictions.
- The overall method maintains a highly efficient self-consistent field optimization algorithm regardless of the sparsity pattern used.
Where Pith is reading between the lines
- The pairing structure could be extended to post-Hartree-Fock methods to introduce controlled locality at higher levels of theory.
- Reaction-specific locality choices might be tuned further to balance speed and accuracy for targeted classes of chemical processes.
- Verification on properties such as molecular geometries or excitation energies would be required to confirm the absence of hidden systematic errors.
- The approach could lower the cost of electronic structure calculations on larger systems where full delocalized orbitals become expensive.
Load-bearing premise
The chosen reaction-matched locality schemes can be imposed without introducing systematic bias that appears only in properties or systems outside the tested reaction energies.
What would settle it
A direct comparison in which the locality schemes produce reaction energies or other properties that differ substantially from standard Hartree-Fock results on a molecule or observable not included in the initial tests.
Figures
read the original abstract
We explore a reorganized framework for the Hartree Fock equations that allows varying patterns of locality to be imposed on the molecular orbitals while maintaining a highly efficient self-consistent field optimization algorithm. Rather than limiting orbitals' spread and then variationally minimizing the energy within those limits, our reorganization neatly pairs each local degree of freedom with a specific solution condition that itself has a naturally local interpretation. These pairs can each be turned on or off, and, regardless of the sparsity pattern used to make such choices, the overall method maintains a fast self-consistent field optimization. We use this structure to test reaction-matched schemes for imposing orbital locality and, through Fock builds that exploit this locality, achieve competitive timings even in modestly sized molecules. Our initial tests also suggest that this approach to imposed orbital locality can be arranged so as to do minimal damage to both Hartree Fock and MP2 reaction energy predictions.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript proposes a reorganized framework for the Hartree-Fock equations that pairs each local degree of freedom with a specific solution condition having a naturally local interpretation. This structure permits turning individual locality constraints on or off while preserving a fast self-consistent field optimization, independent of the chosen sparsity pattern. The authors apply the approach to reaction-matched locality schemes, claiming competitive Fock-build timings for modestly sized molecules and suggesting via initial tests that the imposed locality causes only minimal damage to both HF and MP2 reaction energies.
Significance. If the numerical performance and error claims are substantiated, the reorganization could provide a modular alternative to conventional orbital-localization or density-fitting approximations by decoupling locality choices from the variational procedure. The ability to activate or deactivate individual local pairs without disrupting SCF convergence would be a useful feature for balancing cost and accuracy. The current description, however, supplies no quantitative benchmarks, so the practical significance cannot yet be evaluated.
major comments (2)
- [Abstract] Abstract: the central claim that the method 'can be arranged so as to do minimal damage to both Hartree Fock and MP2 reaction energy predictions' is unsupported by any numerical error tables, mean absolute deviations, error bars, or comparisons to standard local approximations (e.g., local HF, density fitting, or orbital localization). This quantitative evidence is load-bearing for the assertion of practical utility.
- [Abstract] Abstract: the statement that the reorganization 'maintains a fast self-consistent field optimization' 'regardless of the sparsity pattern' is presented without derivation details, pseudocode, convergence analysis, or timing data, leaving the efficiency guarantee unverified and central to the method's advertised advantage.
minor comments (1)
- The abstract would be strengthened by indicating the range of molecular sizes or the specific reaction classes used in the 'initial tests'.
Simulated Author's Rebuttal
We thank the referee for the detailed and constructive report. We respond point-by-point to the major comments below, indicating where we agree that additional support or clarification will strengthen the manuscript.
read point-by-point responses
-
Referee: [Abstract] Abstract: the central claim that the method 'can be arranged so as to do minimal damage to both Hartree Fock and MP2 reaction energy predictions' is unsupported by any numerical error tables, mean absolute deviations, error bars, or comparisons to standard local approximations (e.g., local HF, density fitting, or orbital localization). This quantitative evidence is load-bearing for the assertion of practical utility.
Authors: We agree that the abstract's reference to 'initial tests' would be more persuasive with explicit quantitative support. The body of the manuscript presents reaction-energy results for the chosen locality schemes, but does not include tabulated mean absolute deviations or direct comparisons to other local approximations. In the revised manuscript we will add a dedicated results subsection with error tables, MAD values, and comparisons to standard local HF and orbital-localization approaches to substantiate the claim. revision: yes
-
Referee: [Abstract] Abstract: the statement that the reorganization 'maintains a fast self-consistent field optimization' 'regardless of the sparsity pattern' is presented without derivation details, pseudocode, convergence analysis, or timing data, leaving the efficiency guarantee unverified and central to the method's advertised advantage.
Authors: The reorganized Hartree-Fock framework and the argument that locality constraints can be activated independently without altering the underlying SCF structure are derived in Sections II and III of the manuscript. Nevertheless, we accept that the abstract would benefit from more concrete verification. In the revision we will insert pseudocode for the modified SCF iteration, additional convergence data, and expanded Fock-build timing tables that explicitly vary the sparsity pattern. revision: yes
Circularity Check
Reorganization of HF equations shows no circular derivation
full rationale
The paper presents a reorganized framework for the Hartree-Fock equations that pairs local degrees of freedom with naturally local solution conditions, allowing sparsity patterns to be imposed while preserving fast SCF optimization. This is described as a structural change rather than a fit to data or a self-referential definition. No load-bearing steps reduce by construction to inputs, no fitted parameters are renamed as predictions, and no self-citation chains or ansatzes are invoked to justify the core reorganization. The claims about competitive timings and minimal damage to reaction energies are presented as empirical consequences of the framework, not as tautological outputs of the same inputs. The derivation is therefore self-contained against external benchmarks.
Axiom & Free-Parameter Ledger
Reference graph
Works this paper leans on
-
[1]
, year = 1961, month = jan, journal =
Adams, William H. , year = 1961, month = jan, journal =. On the. doi:10.1063/1.1731622 , urldate =
-
[2]
Ammar, Abdallah and Giner, Emmanuel and Scemama, Anthony , year = 2022, month = sep, journal =. Optimization of. doi:10.1021/acs.jctc.2c00556 , urldate =
-
[3]
Aquilante, Francesco and Boman, Linus and Bostr. Cholesky. Linear-. doi:10.1007/978-90-481-2853-2_13 , urldate =
-
[4]
The Journal of Chemical Physics , volume =
Fast Noniterative Orbital Localization for Large Molecules , author =. The Journal of Chemical Physics , volume =. doi:10.1063/1.2360264 , urldate =
-
[5]
Aroeira, Gustavo J. R. and Davis, Matthew M. and Turney, Justin M. and Schaefer, Henry F. III , year = 2022, month = feb, journal =. Fermi.Jl:. doi:10.1021/acs.jctc.1c00719 , urldate =
-
[6]
The Journal of Chemical Physics , volume =
Quantum. The Journal of Chemical Physics , volume =. doi:10.1063/1.1646356 , urldate =
-
[7]
and Filippi, Claudia , year = 2017, month = nov, journal =
Assaraf, Roland and Moroni, S. and Filippi, Claudia , year = 2017, month = nov, journal =. Optimizing the. doi:10.1021/acs.jctc.7b00648 , urldate =
-
[8]
Assaraf, Roland and Caffarel, Michel , year = 1999, month = dec, journal =. Zero-. doi:10.1103/PhysRevLett.83.4682 , urldate =
-
[9]
Austin, Brian M. and Zubarev, Dmitry Yu. and Lester, William A. Jr. , year = 2012, month = jan, journal =. Quantum. doi:10.1021/cr2001564 , urldate =
-
[10]
The Journal of Chemical Physics , volume =
An Energy Decomposition Analysis for Intermolecular Interactions from an Absolutely Localized Molecular Orbital Reference at the Coupled-Cluster Singles and Doubles Level , author =. The Journal of Chemical Physics , volume =. doi:10.1063/1.3674992 , urldate =
-
[11]
The Journal of Chemical Physics , volume =
Chebyshev Expansion Methods for Electronic Structure Calculations on Large Molecular Systems , author =. The Journal of Chemical Physics , volume =. doi:10.1063/1.474158 , urldate =
-
[12]
Vanadium Oxide Compounds with Quantum
Bande, Annika and L. Vanadium Oxide Compounds with Quantum. Physical Chemistry Chemical Physics , volume =. doi:10.1039/B803571G , urldate =
-
[13]
Reviews of Modern Physics , volume =
Coupled-Cluster Theory in Quantum Chemistry , author =. Reviews of Modern Physics , volume =. doi:10.1103/RevModPhys.79.291 , urldate =
-
[14]
Correlated Natural Transition Orbital Framework for Low-Scaling Excitation Energy Calculations (
Baudin, Pablo and Kristensen, Kasper , year = 2017, month = jun, journal =. Correlated Natural Transition Orbital Framework for Low-Scaling Excitation Energy Calculations (. doi:10.1063/1.4984820 , urldate =
-
[15]
, year = 1980, month = jan, journal =
Bauschlicher, Jr., Charles W. , year = 1980, month = jan, journal =. The Construction of Modified Virtual Orbitals (. doi:10.1063/1.439243 , urldate =
-
[16]
Physical Chemistry Chemical Physics , volume =
Spurious Proton Transfer in Hydrogen Bonded Dimers , author =. Physical Chemistry Chemical Physics , volume =. doi:10.1039/D4CP00907J , urldate =
-
[17]
Becca, Federico and Sorella, Sandro , year = 2017, publisher =. Quantum. doi:10.1017/9781316417041 , urldate =
-
[18]
Benedek, N. A. and Snook, I. K. and Towler, M. D. and Needs, R. J. , year = 2006, month = sep, journal =. Quantum. doi:10.1063/1.2338032 , urldate =
-
[19]
The Journal of Chemical Physics , volume =
Orbital Pair Selection for Relative Energies in the Domain-Based Local Pair Natural Orbital Coupled-Cluster Method , author =. The Journal of Chemical Physics , volume =. doi:10.1063/5.0100010 , urldate =
-
[20]
Physical Chemistry Chemical Physics , volume =
Approaching Closed-Shell Accuracy for Radicals Using Coupled Cluster Theory with Perturbative Triple Substitutions , author =. Physical Chemistry Chemical Physics , volume =. doi:10.1039/B304542K , urldate =
-
[21]
Systematic Lowering of the Scaling of
Bienvenu, Antoine and Feldt, Jonas and Toulouse, Julien and Assaraf, Roland , year = 2022, month = aug, journal =. Systematic Lowering of the Scaling of. doi:10.1103/PhysRevE.106.025301 , urldate =
-
[22]
and Gu, Feng Long and Kirtman, Bernard , year = 2001, month = may, journal =
Bishop, David M. and Gu, Feng Long and Kirtman, Bernard , year = 2001, month = may, journal =. Coupled-Perturbed. doi:10.1063/1.1356019 , urldate =
-
[23]
Stein, Christopher , year = 2024, journal =
Bogo, Nicola and J. Stein, Christopher , year = 2024, journal =. Benchmarking. doi:10.1039/D4CP01866D , urldate =
-
[24]
Towards an Exact Description of Electronic Wavefunctions in Real Solids , author =. Nature , volume =. doi:10.1038/nature11770 , urldate =
-
[25]
Booth, George H. and Thom, Alex J. W. and Alavi, Ali , year = 2009, month = aug, journal =. Fermion. doi:10.1063/1.3193710 , urldate =
-
[26]
Bostr. Coupled. Journal of Chemical Theory and Computation , volume =. doi:10.1021/ct3003018 , urldate =
-
[27]
and Pulay, Peter , year = 1993, journal =
Boughton, James W. and Pulay, Peter , year = 1993, journal =. Comparison of the Boys and. doi:10.1002/jcc.540140615 , urldate =
-
[28]
Configuration Interaction with
Bou. Configuration Interaction with. Chemical Physics Letters , volume =. doi:10.1016/S0009-2614(01)00896-X , urldate =
-
[29]
The Journal of Chemical Physics , volume =
Elucidating the Molecular Orbital Dependence of the Total Electronic Energy in Multireference Problems , author =. The Journal of Chemical Physics , volume =. doi:10.1063/5.0090342 , urldate =
-
[30]
Buckingham, A. D. , year = 1967, pages =. Permanent and. Advances in. doi:10.1002/9780470143582.ch2 , urldate =
-
[31]
Numerische Mathematik , volume =
Rank-One Modification of the Symmetric Eigenproblem , author =. Numerische Mathematik , volume =. doi:10.1007/BF01396012 , urldate =
-
[32]
Bunge, C. F. and Barrientos, J. A. and Bunge, A. V. , year = 1993, month = jan, journal =. Roothaan-. doi:10.1006/adnd.1993.1003 , urldate =
-
[33]
Burns, Lori A. and Faver, John C. and Zheng, Zheng and Marshall, Michael S. and Smith, Daniel G. A. and Vanommeslaeghe, Kenno and MacKerell, Jr., Alexander D. and Merz, Jr., Kenneth M. and Sherrill, C. David , year = 2017, month = sep, journal =. The. doi:10.1063/1.5001028 , urldate =
-
[34]
, year = 2016, month = jul, journal =
Busemeyer, Brian and Dagrada, Mario and Sorella, Sandro and Casula, Michele and Wagner, Lucas K. , year = 2016, month = jul, journal =. Competing Collinear Magnetic Structures in Superconducting. doi:10.1103/PhysRevB.94.035108 , urldate =
-
[35]
Solving the Quantum Many-Body Problem with Artificial Neural Networks , author =. Science , volume =. doi:10.1126/science.aag2302 , urldate =
-
[36]
Reviews of Modern Physics , volume =
The Electronic Properties of Graphene , author =. Reviews of Modern Physics , volume =. doi:10.1103/RevModPhys.81.109 , urldate =
-
[37]
Correlated Geminal Wave Function for Molecules:
Casula, Michele and Attaccalite, Claudio and Sorella, Sandro , year = 2004, month = oct, journal =. Correlated Geminal Wave Function for Molecules:. doi:10.1063/1.1794632 , urldate =
-
[38]
Ceperley, D. M. and Alder, B. J. , year = 1980, month = aug, journal =. Ground. doi:10.1103/PhysRevLett.45.566 , urldate =
-
[39]
Nature Reviews Physics , author =
Variational Quantum Algorithms , author =. Nature Reviews Physics , volume =. doi:10.1038/s42254-021-00348-9 , urldate =
-
[40]
Chaitanya, K. V. S. Shiv and Bambah, B. A. , year = 2018, month = jan, number =. A. doi:10.48550/arXiv.1801.00544 , urldate =. arXiv , keywords =:1801.00544 , primaryclass =
work page internal anchor Pith review Pith/arXiv arXiv doi:10.48550/arxiv.1801.00544 2018
-
[41]
Champagne, Beno. Calculations of. Advances in. doi:10.1002/0471428019.ch2 , urldate =
-
[42]
Chaquin, Patrick and Canac, Yves and Lepetit, Christine and Zargarian, Davit and Chauvin, Remi , year = 2016, journal =. Estimating Local Bonding/Antibonding Character of Canonical Molecular Orbitals from Their Energy Derivatives. doi:10.1002/qua.25174 , urldate =
-
[43]
The Journal of Chemical Physics , volume =
Discrete Energy Representation and Generalized Propagation of Physical Systems , author =. The Journal of Chemical Physics , volume =. doi:10.1063/1.476017 , urldate =
-
[44]
The Journal of Chemical Physics , volume =
A General and Efficient Filter-diagonalization Method without Time Propagation , author =. The Journal of Chemical Physics , volume =. doi:10.1063/1.471997 , urldate =
-
[45]
A Theoretical Investigation of the Structure and Vibrational Frequencies of
Cho, Han-Gook and Cheong, Byeong-Sea , year = 2000, month = jan, journal =. A Theoretical Investigation of the Structure and Vibrational Frequencies of. doi:10.1016/S0166-1280(99)00183-9 , urldate =
-
[46]
Chowdhury, Sujaul , editor =. Variational. Monte. doi:10.1007/978-3-031-23294-7_5 , urldate =
-
[47]
Clark, Bryan K. and Morales, Miguel A. and McMinis, Jeremy and Kim, Jeongnim and Scuseria, Gustavo E. , year = 2011, month = dec, journal =. Computing the Energy of a Water Molecule Using Multideterminants:. doi:10.1063/1.3665391 , urldate =
-
[48]
The Journal of Chemical Physics , volume =
An Excitation Matched Local Correlation Approach to Excited State Specific Perturbation Theory , author =. The Journal of Chemical Physics , volume =. doi:10.1063/5.0280479 , urldate =
-
[49]
Clune, Rachel and Shea, Jacqueline A. R. and Neuscamman, Eric , year = 2020, month = oct, journal =. N5-. doi:10.1021/acs.jctc.0c00308 , urldate =
-
[50]
Couty, Marc and Bayse, Craig A. and Hall, Michael B. , year = 1997, month = oct, journal =. Extremely Localized Molecular Orbitals (. doi:10.1007/s002140050242 , urldate =
-
[51]
Daniel and Schaefer III, Henry F
Crawford, T. Daniel and Schaefer III, Henry F. , year = 2000, pages =. An. Reviews in. doi:10.1002/9780470125915.ch2 , urldate =
-
[53]
Cremer, Dieter , year = 2011, journal =. M ller--. doi:10.1002/wcms.58 , urldate =
-
[54]
Csonka, G. Evaluation of. Journal of Chemical Theory and Computation , volume =. doi:10.1021/ct8004479 , urldate =
-
[55]
Cullen, John M. and Zerner, Michael C. , year = 1982, month = oct, journal =. The Linked Singles and Doubles Model:. doi:10.1063/1.444319 , urldate =
-
[56]
Advances in Physics , volume =
Atomic Polarizabilities and Shielding Factors , author =. Advances in Physics , volume =. doi:10.1080/00018736200101302 , urldate =
-
[57]
Damour, Yann and Scemama, Anthony and Jacquemin, Denis and Kossoski, F. State-. Journal of Chemical Theory and Computation , volume =. doi:10.1021/acs.jctc.4c00034 , urldate =
-
[58]
Dash, Monika and Feldt, Jonas and Moroni, Saverio and Scemama, Anthony and Filippi, Claudia , year = 2019, month = sep, journal =. Excited. doi:10.1021/acs.jctc.9b00476 , urldate =
-
[59]
The Journal of Chemical Physics , volume =
Analytic Energy Derivatives for the Calculation of the First-Order Molecular Properties Using the Domain-Based Local Pair-Natural Orbital Coupled-Cluster Theory , author =. The Journal of Chemical Physics , volume =. doi:10.1063/1.4962369 , urldate =
-
[60]
David, Rolf and Tu. Competing. Journal of the American Chemical Society , volume =. doi:10.1021/jacs.4c03445 , urldate =
-
[61]
Dereka, Bogdan and Lewis, Nicholas H. C. and Keim, Jonathan H. and Snyder, Scott A. and Tokmakoff, Andrei , year = 2022, month = jan, journal =. Characterization of. doi:10.1021/acs.jpcb.1c09572 , urldate =
-
[62]
Dereka, Bogdan and Lewis, Nicholas H. C. and Zhang, Yong and Hahn, Nathan T. and Keim, Jonathan H. and Snyder, Scott A. and Maginn, Edward J. and Tokmakoff, Andrei , year = 2022, month = may, journal =. Exchange-. doi:10.1021/jacs.2c00154 , urldate =
-
[64]
Birgitta and Lin, Lin , year = 2021, month = aug, doi =
Dong, Yulong and Whaley, K. Birgitta and Lin, Lin , year = 2021, month = aug, doi =. A
2021
-
[65]
Dreuw, Andreas and. Failure of. Journal of the American Chemical Society , volume =. doi:10.1021/ja039556n , urldate =
-
[66]
The Journal of Chemical Physics , volume =
Long-Range Charge-Transfer Excited States in Time-Dependent Density Functional Theory Require Non-Local Exchange , author =. The Journal of Chemical Physics , volume =. doi:10.1063/1.1590951 , urldate =
-
[67]
Dreuw, Andreas and. Single-. Chemical Reviews , volume =. doi:10.1021/cr0505627 , urldate =
-
[68]
, year = 2007, month = aug, journal =
Drukarev, Evgenii G. , year = 2007, month = aug, journal =. Atomic Physics: Computer Calculations and Theoretical Analysis , shorttitle =. doi:10.1070/PU2007v050n08ABEH006372 , urldate =
-
[69]
Gaussian basis sets for use in correlated molecular calculations
Dunning, Jr., Thom H. , year = 1989, month = jan, journal =. Gaussian Basis Sets for Use in Correlated Molecular Calculations. doi:10.1063/1.456153 , urldate =
-
[70]
Dykstra, Clifford E. and Jasien, Paul G. , year = 1984, month = aug, journal =. Derivative. doi:10.1016/0009-2614(84)85607-9 , urldate =
-
[71]
Direct Observation of Chirality-Induced Spin Selectivity in Electron Donor--Acceptor Molecules , author =. Science , volume =. doi:10.1126/science.adj5328 , urldate =
-
[72]
Ehlert, Sebastian and Grimme, Stefan and Hansen, Andreas , year = 2022, month = jun, journal =. Conformational. doi:10.1021/acs.jpca.2c02439 , urldate =
-
[73]
Entwistle, M. T. and Sch. Electronic Excited States in Deep Variational. Nature Communications , volume =. doi:10.1038/s41467-022-35534-5 , urldate =
-
[74]
Epifanovsky, Evgeny and Gilbert, Andrew T. B. and Feng, Xintian and Lee, Joonho and Mao, Yuezhi and Mardirossian, Narbe and Pokhilko, Pavel and White, Alec F. and Coons, Marc P. and Dempwolff, Adrian L. and Gan, Zhengting and Hait, Diptarka and Horn, Paul R. and Jacobson, Leif D. and Kaliman, Ilya and Kussmann, J. Software for the Frontiers of Quantum Che...
-
[75]
and Lin, Lin and Nakatsukasa, Yuji , year = 2021, month = oct, number =
Epperly, Ethan N. and Lin, Lin and Nakatsukasa, Yuji , year = 2021, month = oct, number =. A. arXiv , keywords =:2110.07492 , primaryclass =
arXiv 2021
-
[76]
and Lin, Lin and Nakatsukasa, Yuji , year = 2021, month = oct, number =
Epperly, Ethan N. and Lin, Lin and Nakatsukasa, Yuji , year = 2021, month = oct, number =. A. doi:10.48550/arXiv.2110.07492 , urldate =. arXiv , keywords =:2110.07492 , primaryclass =
-
[77]
and Lin, Lin and Nakatsukasa, Yuji , year = 2022, month = sep, journal =
Epperly, Ethan N. and Lin, Lin and Nakatsukasa, Yuji , year = 2022, month = sep, journal =. A. doi:10.1137/21M145954X , urldate =
-
[78]
Evers et al, Theory of Chirality Induced Spin Selectivity: Progress and Challenges, Adv
Evers, Ferdinand and Aharony, Amnon and. Theory of. Advanced Materials , volume =. doi:10.1002/adma.202106629 , urldate =
-
[79]
Excited States in Variational
-
[80]
Fang, Zongtang and Vasiliu, Monica and Peterson, Kirk A. and Dixon, David A. , year = 2017, month = mar, journal =. Prediction of. doi:10.1021/acs.jctc.6b00971 , urldate =
-
[81]
Fang, Zongtang and Lee, Zachary and Peterson, Kirk A. and Dixon, David A. , year = 2016, month = aug, journal =. Use of. doi:10.1021/acs.jctc.6b00327 , urldate =
-
[82]
, year = 2021, month = feb, journal =
Fay, Thomas P. , year = 2021, month = feb, journal =. Chirality-. doi:10.1021/acs.jpclett.1c00009 , urldate =
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.