Do Larger Models Really Win in Drug Discovery? A Benchmark Assessment of Model Scaling in AI-Driven Molecular Property and Activity Prediction
Pith reviewed 2026-05-19 17:37 UTC · model grok-4.3
The pith
Classical ML models outperform larger pretrained and LLM approaches in most molecular prediction tasks for drug discovery
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
Across 78 endpoint and split entries for molecular properties, toxicity, safety liabilities and biological activity, classical ML such as RF(ECFP4) and ExtraTrees(RDKit) win 116 out of 156 fold mean comparisons, GNNs such as GIN and Ligandformer win 25, pretrained sequence models such as MoLFormer and ChemBERTa2 win 12, and LLM based SAR baselines win three. ML dominates random split interpolation but loses part of this advantage under harder splits; GNN and sequence models also decline but gain relative ground, whereas LLM based SAR is weaker in absolute terms yet less sensitive to the split axis. SAR knowledge derived from training folds improves many GPT5.5-SAR and Opus4.7-SAR metrics but
What carries the argument
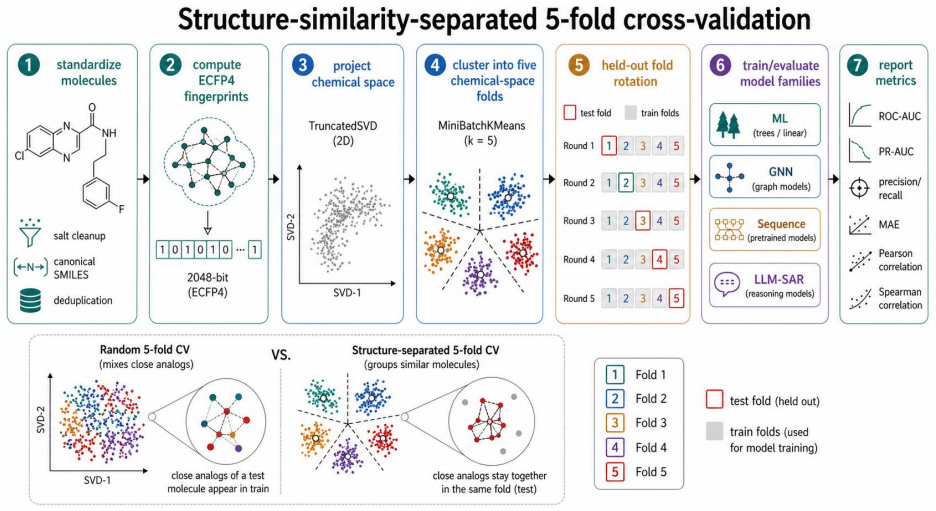
The tiered cross-validation benchmark using random, Murcko scaffold and structure-separated 5-fold splits on 26 endpoints grouped into ADME, toxicity and bioactivity classes to compare four model families under increasing generalization demands
If this is right
- Classical ML models achieve highest accuracy on easier random splits but their lead narrows on scaffold and structure-separated splits
- GNNs and pretrained sequence models lose ground in absolute terms on harder splits yet improve their relative ranking against classical ML
- LLM-based SAR baselines deliver lower absolute performance but remain more stable when split difficulty increases
- Incorporating SAR knowledge from the training folds raises LLM metrics without turning rule-based reasoning into a universal replacement for supervised predictors
- Overall success depends on the fit between model family, task type and validation scenario rather than on model scale
Where Pith is reading between the lines
- Teams running routine high-throughput screens could default to fast classical models to reduce compute costs without sacrificing accuracy
- LLMs may still add value in very low-data regimes for generating SAR hypotheses even if they trail on raw prediction metrics
- Future work should test these patterns on time-based or truly prospective splits to check whether the observed family rankings hold in live discovery campaigns
Load-bearing premise
The 78 endpoint and split entries using random, Murcko scaffold and structure-separated 5-fold CV adequately represent the spectrum of real-world drug discovery challenges from closed-library retrospective evaluation to novel chemotype library expansion
What would settle it
A follow-up study on a fresh set of endpoints or on prospectively collected compounds where larger pretrained or LLM models consistently beat classical ML across all three split types would show the scaling assumption holds after all
Figures




read the original abstract
The rapid growth of molecular foundation models and large language models has encouraged a scale centred view of AI in drug discovery, in which larger pretrained models are expected to supersede compact cheminformatics models and graph neural networks (GNNs) trained for individual tasks. We test this assumption across 26 endpoints for molecular properties, toxicity, safety liabilities and biological activity, grouped into ADME, toxicity and bioactivity classes. The benchmark contains 78 endpoint and split entries spanning random, Murcko scaffold and structure separated 5-fold CV. Ordered from easiest to hardest, these splits approximate retrospective evaluation on a closed library, scaffold expansion in hit to lead, and library expansion on novel chemotypes. Each entry includes ML, GNN, pretrained molecular sequence and LLM based SAR families. Across 156 fold mean comparisons, classical ML such as RF(ECFP4) and ExtraTrees(RDKit) win 116, GNNs such as GIN and Ligandformer win 25, pretrained sequence models such as MoLFormer and ChemBERTa2 win 12, and LLM based SAR baselines win three. ML dominates random split interpolation but loses part of this advantage under harder splits; GNN and sequence models also decline but gain relative ground, whereas LLM based SAR is weaker in absolute terms yet less sensitive to the split axis. Paired bootstrap analyses support family level trends more strongly than individual model rankings. SAR knowledge derived from training folds improves many GPT5.5-SAR and Opus4.7-SAR metrics but does not make rule based reasoning a universal substitute for supervised predictors. Compact specialized models remain highly effective for molecular property and activity prediction. Larger models add value for SAR interpretation and reasoning in low data settings, but predictive performance depends on the fit among model, task and validation scenario, not on scale alone.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript benchmarks classical ML models (e.g., RF(ECFP4), ExtraTrees(RDKit)), GNNs (e.g., GIN, Ligandformer), pretrained sequence models (e.g., MoLFormer, ChemBERTa2), and LLM-based SAR baselines across 26 endpoints in ADME, toxicity, and bioactivity categories. Using 78 endpoint/split entries with random, Murcko scaffold, and structure-separated 5-fold CV, it reports classical ML winning 116 of 156 fold-mean comparisons, GNNs winning 25, pretrained models 12, and LLMs 3. The central claim is that compact specialized models remain highly effective for predictive performance, while larger models add value mainly for SAR interpretation in low-data settings, with performance depending on model-task-validation fit rather than scale alone.
Significance. If the empirical results hold under scrutiny, the work is significant for providing a large-scale, multi-family comparison that challenges scale-centric assumptions in AI for drug discovery. It supplies concrete win-rate data and notes relative robustness of LLM-SAR to split difficulty, which could inform model selection. The use of paired bootstrap analyses for family trends and the ordering of splits by difficulty are positive features that increase the benchmark's reference value.
major comments (2)
- [Results] Results section (win-count reporting): The abstract and results state classical ML wins 116 of 156 comparisons, yet no per-family variance, confidence intervals, or explicit tie-handling rules are supplied alongside the bootstrap analyses; without these, the strength of the family-level dominance claim cannot be fully evaluated from the numbers alone.
- [Methods] Methods (validation splits): The structure-separated 5-fold CV is used to approximate novel-chemotype library expansion, but the manuscript provides no quantitative checks for residual chemical similarity or analog leakage between folds, nor any comparison against strict temporal splits by assay or patent date; this leaves open whether the observed classical-ML advantage would persist under the distribution shifts typical of prospective drug-discovery validation.
minor comments (2)
- [Abstract] Abstract: The terms 'GPT5.5-SAR' and 'Opus4.7-SAR' appear without prior definition or reference to the underlying LLM versions and prompting strategy.
- [Tables] Tables: Ensure every results table lists the exact number of comparisons contributing to each win count so readers can verify the 156 total and the per-class breakdowns.
Simulated Author's Rebuttal
We thank the referee for the constructive comments and for recognizing the benchmark's scope and reference value. We address each major comment below with clarifications and indicate where revisions will be made.
read point-by-point responses
-
Referee: [Results] Results section (win-count reporting): The abstract and results state classical ML wins 116 of 156 comparisons, yet no per-family variance, confidence intervals, or explicit tie-handling rules are supplied alongside the bootstrap analyses; without these, the strength of the family-level dominance claim cannot be fully evaluated from the numbers alone.
Authors: The win counts are presented as descriptive aggregates, while the paired bootstrap analyses were used to evaluate family-level trends. We agree that explicit reporting of per-family variance, confidence intervals, and tie-handling rules would strengthen interpretability. In the revised manuscript we will add bootstrap-derived confidence intervals for each family's win rate and specify the tie-handling procedure (ties assigned proportionally to the tied families). revision: yes
-
Referee: [Methods] Methods (validation splits): The structure-separated 5-fold CV is used to approximate novel-chemotype library expansion, but the manuscript provides no quantitative checks for residual chemical similarity or analog leakage between folds, nor any comparison against strict temporal splits by assay or patent date; this leaves open whether the observed classical-ML advantage would persist under the distribution shifts typical of prospective drug-discovery validation.
Authors: We will incorporate quantitative checks for residual similarity, including mean and distribution of ECFP4 Tanimoto similarities between training and test folds, to document the degree of analog leakage. However, the source datasets do not contain consistent assay or patent dates for all 26 endpoints, precluding a uniform temporal-split comparison. We will note this data limitation explicitly and discuss the structure-separated split as a practical proxy for prospective validation. revision: partial
- Direct comparison to strict temporal splits by assay or patent date, because consistent temporal metadata is unavailable across the full set of public datasets used.
Circularity Check
No circularity: empirical benchmark rests on direct held-out comparisons
full rationale
The paper reports model performance via explicit 5-fold CV on 78 endpoint/split combinations, counting wins across 156 comparisons and supporting trends with paired bootstrap. No equations, fitted parameters, or derivations are present; results are computed directly from held-out test folds rather than being redefined or predicted from the training statistics themselves. No self-citations are invoked as load-bearing uniqueness theorems or ansatzes. The central claim therefore remains an independent empirical observation rather than a self-referential reduction.
Axiom & Free-Parameter Ledger
axioms (1)
- domain assumption The random, Murcko scaffold, and structure-separated 5-fold CV splits approximate retrospective closed-library evaluation, scaffold expansion in hit-to-lead, and library expansion on novel chemotypes.
Reference graph
Works this paper leans on
-
[1]
Feinberg, Joseph Gomes, Caleb Geniesse, Aneesh S
Zhenqin Wu, Bharath Ramsundar, Evan N. Feinberg, Joseph Gomes, Caleb Geniesse, Aneesh S. Pappu, Karl Leswing, and Vijay Pande. Moleculenet: A benchmark for molec- ular machine learning.Chemical Science, 9:513–530, 2018. doi: 10.1039/C7SC02664A
-
[2]
Kexin Huang, Tianfan Fu, Wenhao Gao, Yue Zhao, Yusuf Roohani, Jure Leskovec, Connor W. Coley, Cao Xiao, Jimeng Sun, and Marinka Zitnik. Therapeutics data commons: Machine learning datasets and tasks for drug discovery and development. InProceedings of the Neural Information Processing Systems Track on Datasets and Benchmarks, 2021. URLhttps:// arxiv.org/a...
-
[3]
Megan Stanley, John F. Bronskill, Krzysztof Maziarz, Henryk Misztela, Julien Lanini, Marwin Segler, Nadine Schneider, and Marc Brockschmidt. Fs-mol: A few-shot learning dataset of molecules. InNeurIPS Datasets and Benchmarks Track, 2021. URLhttps://openreview. net/forum?id=701FtuyLlAd
work page 2021
-
[4]
Ana Laura Dias, Latimah Bustillo, and Tiago Rodrigues. Limitations of representation learning in small molecule property prediction.Nature Communications, 14:6394, 2023. doi: 10.1038/ s41467-023-41967-3. URLhttps://www.nature.com/articles/s41467-023-41967-3
work page 2023
- [5]
-
[6]
Gintautas Kamuntavicius, Tanya Paquet, Orestis Bastas, Dainius Salkauskas, Alvaro Prat, Hisham Abdel Aty, Aurimas Pabrinkis, Povilas Norvaisas, and Roy Tal. Benchmarking ma- chine learning in admet predictions: The practical impact of feature representations in ligand- based models.Journal of Cheminformatics, 17:108, 2025. doi: 10.1186/s13321-025-01041-0....
-
[7]
ChemBERTa: large -scale self -supervised pretraining fo r molecular property prediction
Seyone Chithrananda, Gabriel Grand, and Bharath Ramsundar. Chemberta: Large-scale self- supervised pretraining for molecular property prediction, 2020. URLhttps://arxiv.org/ abs/2010.09885
-
[8]
ChemBERTa- 2: Towards chemical foundation models.arXiv preprint arXiv:2209.01712, 2022
Walid Ahmad, Eric Simon, Seyone Chithrananda, Gabriel Grand, and Bharath Ramsundar. Chemberta-2: Towards chemical foundation models, 2022. URLhttps://arxiv.org/abs/ 2209.01712
-
[9]
Jerret Ross, Brian Belgodere, Vijil Chenthamarakshan, Inkit Padhi, Youssef Mroueh, and Payel Das. Large-scale chemical language representations capture molecular structure and properties.Nature Machine Intelligence, 2022. URLhttps://www.nature.com/articles/ s42256-022-00580-7
work page 2022
-
[10]
Raymond R. Tice, Christopher P. Austin, Robert J. Kavlock, and John R. Bucher. Tox21 challenge to build predictive models of nuclear receptor and stress response pathways as medi- ated by exposure to environmental chemicals and drugs.Frontiers in Environmental Science, 16
-
[11]
URLhttps://www.frontiersin.org/journals/environmental-science/articles/ 10.3389/fenvs.2015.00085/full
-
[12]
Andreas Mayr, Gunter Klambauer, Thomas Unterthiner, and Sepp Hochreiter. Qsar modeling of tox21 challenge stress response and nuclear receptor signaling toxicity assays.Frontiers in Environmental Science, 2016. URLhttps://www.frontiersin.org/articles/10.3389/ fenvs.2016.00003/full
-
[13]
Poonam Chitale, Alexander D. Lemenze, Emily C. Fogarty, Avi Shah, Courtney Grady, Aubrey R. Odom-Mabey, W. Evan Johnson, Jason H. Yang, A. Murat Eren, Roland Brosch, Pradeep Kumar, and David Alland. A comprehensive update to the mycobac- terium tuberculosis h37rv reference genome.Nature Communications, 13:7068, 2022. doi: 10.1038/s41467-022-34853-x
-
[14]
Wallace, Vineet Kumar, Ursula Pieper, Andrej Sali, Jeremy R
Francisco Mart ’inez-Jim ’enez, George Papadatos, Li Yang, Iain M. Wallace, Vineet Kumar, Ursula Pieper, Andrej Sali, Jeremy R. Brown, John P. Overington, and Marc A. Marti-Renom. Target prediction for an open access set of compounds active against mycobacterium tuberculosis.PLoS Computational Biology, 9(10):e1003253, 2013. doi: 10.1371/journal.pcbi.1003253
-
[15]
Thomas Lane, Daniel P. Russo, Kimberley M. Zorn, Alex M. Clark, Alexandru Korotcov, Valery Tkachenko, Robert C. Reynolds, Alexander L. Perryman, Joel S. Freundlich, and Sean Ekins. Comparing and validating machine learning models for mycobacterium tuber- culosis drug discovery.Molecular Pharmaceutics, 15(10):4346–4360, 2018. doi: 10.1021/acs. molpharmaceu...
work page doi:10.1021/acs 2018
-
[16]
Shiroh Iwanaga, Rie Kubota, Tsubasa Nishi, Sumalee Kamchonwongpaisan, Somdet Srichairatanakool, Naoaki Shinzawa, Din Syafruddin, Masao Yuda, and Chairat Uthaipibull. Genome-wide functional screening of drug-resistance genes in plasmodium falciparum.Nature Communications, 13:6163, 2022. doi: 10.1038/s41467-022-33804-w
-
[17]
Cluster Computing 6(3), 215–226 (Jul 2003), https://doi.org/10.1023/A: 1023588520138
Leo Breiman. Random forests.Machine Learning, 45(1):5–32, 2001. doi: 10.1023/A: 1010933404324
work page doi:10.1023/a: 2001
-
[18]
Extremely randomized trees.Machine Learning, 63(1):3–42, 2006
Pierre Geurts, Damien Ernst, and Louis Wehenkel. Extremely randomized trees.Machine Learning, 63(1):3–42, 2006. doi: 10.1007/s10994-006-6226-1
-
[19]
Jerome H. Friedman. Greedy function approximation: A gradient boosting machine.The Annals of Statistics, 29(5):1189–1232, 2001. doi: 10.1214/aos/1013203451
-
[20]
Corinna Cortes and Vladimir Vapnik. Support-vector networks.Machine Learning, 20:273– 297, 1995. doi: 10.1007/BF00994018
-
[21]
Tianqi Chen and Carlos Guestrin. Xgboost: A scalable tree boosting system. InProceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, pages 785–794, 2016. doi: 10.1145/2939672.2939785
-
[22]
Extended-connectivity fingerprints.J
David Rogers and Mathew Hahn. Extended-connectivity fingerprints.Journal of Chemical Information and Modeling, 50(5):742–754, 2010. doi: 10.1021/ci100050t. 17
-
[23]
Sereina Riniker and Gregory A. Landrum. Similarity maps – a visualization strategy for molecular fingerprints and machine-learning methods.Journal of Cheminformatics, 5:43, 2013. doi: 10.1186/1758-2946-5-43
-
[24]
Justin Gilmer, Samuel S. Schoenholz, Patrick F. Riley, Oriol Vinyals, and George E. Dahl. Neural message passing for quantum chemistry. InProceedings of the 34th International Con- ference on Machine Learning, volume 70 ofProceedings of Machine Learning Research, pages 1263–1272, 2017. URLhttps://proceedings.mlr.press/v70/gilmer17a.html
work page 2017
-
[25]
Xiaomin Fang, Lihang Liu, et al. Geometric deep learning for molecular property prediction: A review.Nature Machine Intelligence, 2023
work page 2023
-
[26]
Semi-Supervised Classification with Graph Convolutional Networks
Thomas N. Kipf and Max Welling. Semi-supervised classification with graph convolutional networks. InInternational Conference on Learning Representations, 2017. URLhttps:// arxiv.org/abs/1609.02907
work page internal anchor Pith review Pith/arXiv arXiv 2017
-
[27]
Petar Velickovic, Guillem Cucurull, Arantxa Casanova, Adriana Romero, Pietro Lio, and Yoshua Bengio. Graph attention networks. InInternational Conference on Learning Repre- sentations, 2018. URLhttps://arxiv.org/abs/1710.10903
work page internal anchor Pith review Pith/arXiv arXiv 2018
-
[28]
How Powerful are Graph Neural Networks?
Keyulu Xu, Weihua Hu, Jure Leskovec, and Stefanie Jegelka. How powerful are graph neural networks? InInternational Conference on Learning Representations, 2019. URLhttps: //arxiv.org/abs/1810.00826
work page internal anchor Pith review Pith/arXiv arXiv 2019
-
[29]
Ligandformer: A Graph Neural Network for Predicting Compound Property with Robust Interpretation
Jinjiang Guo, Qi Liu, Han Guo, and Xi Lu. Ligandformer: A graph neural network for predicting compound property with robust interpretation, 2022. URLhttps://arxiv.org/ abs/2202.10873
work page internal anchor Pith review Pith/arXiv arXiv 2022
-
[30]
Chawla, Olaf Wiest, and Xiangliang Zhang
Taicheng Guo, Kehan Guo, Bozhao Nan, Zixing Liang, Zhichun Guo, Nitesh V. Chawla, Olaf Wiest, and Xiangliang Zhang. What can large language models do in chemistry? a comprehensive benchmark on eight tasks.arXiv preprint arXiv:2305.18365, 2023. doi: 10. 48550/arXiv.2305.18365
-
[31]
David Weininger. Smiles, a chemical language and information system. 1. introduction to methodology and encoding rules.Journal of Chemical Information and Computer Sciences, 28(1):31–36, 1988. doi: 10.1021/ci00057a005
-
[32]
Guy W. Bemis and Mark A. Murcko. The properties of known drugs. 1. molecular frameworks. Journal of Medicinal Chemistry, 39(15):2887–2893, 1996. doi: 10.1021/jm9602928
-
[33]
Best practices for qsar model development, validation, and exploitation
Alexander Tropsha. Best practices for qsar model development, validation, and exploitation. Molecular Informatics, 29(6–7):476–488, 2010. doi: 10.1002/minf.201000061
-
[34]
Jos ’e Jim ’enez-Luna, Francesca Grisoni, and Gisbert Schneider. Drug discovery with explain- able artificial intelligence.Nature Machine Intelligence, 2:573–584, 2020. doi: 10.1038/ s42256-020-00236-4. 18
work page 2020
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.