Quantum annealing for materials
Pith reviewed 2026-06-28 09:25 UTC · model grok-4.3
The pith
Path-integral molecular dynamics implements quantum annealing to minimize material potential energy surfaces without explicit wavefunction manipulation.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
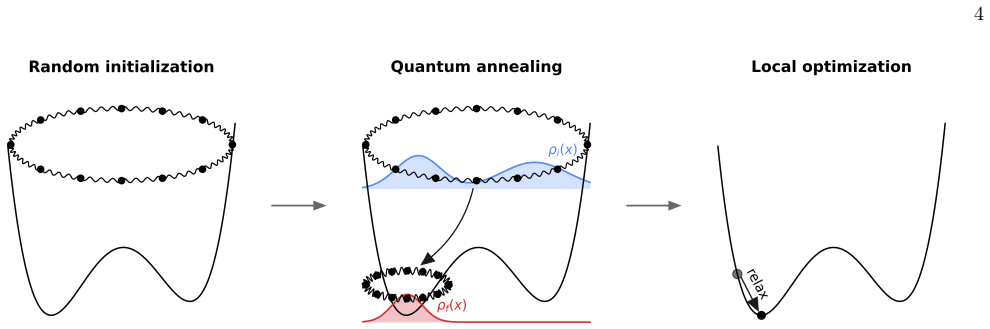
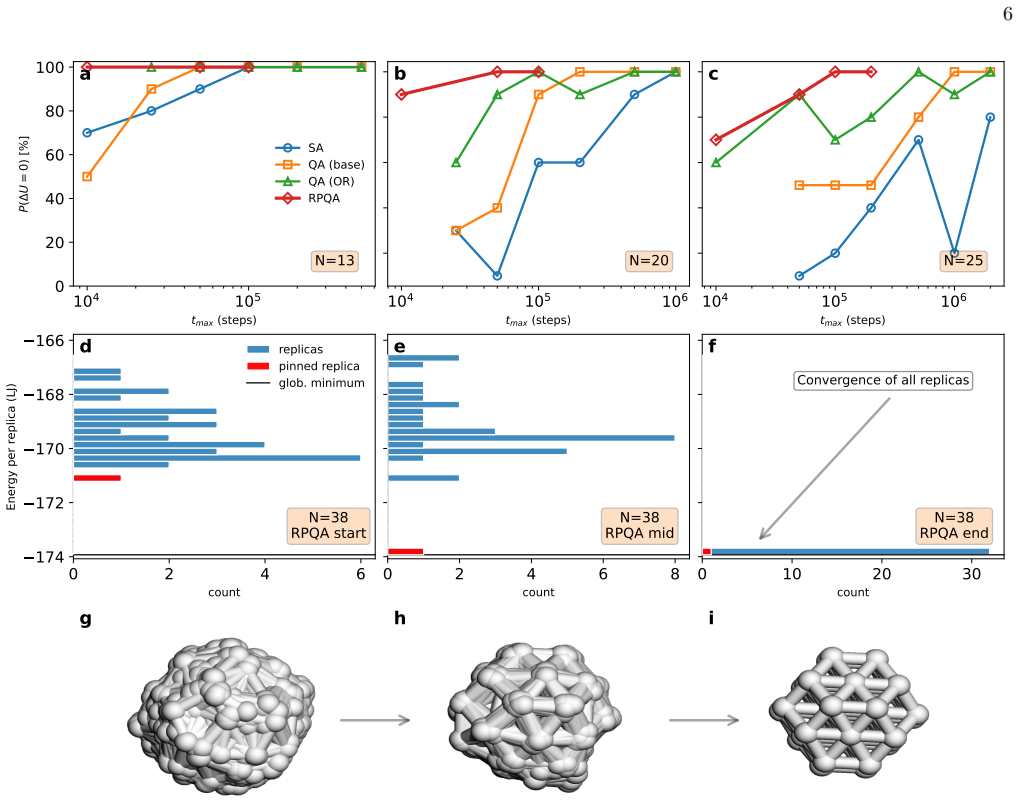
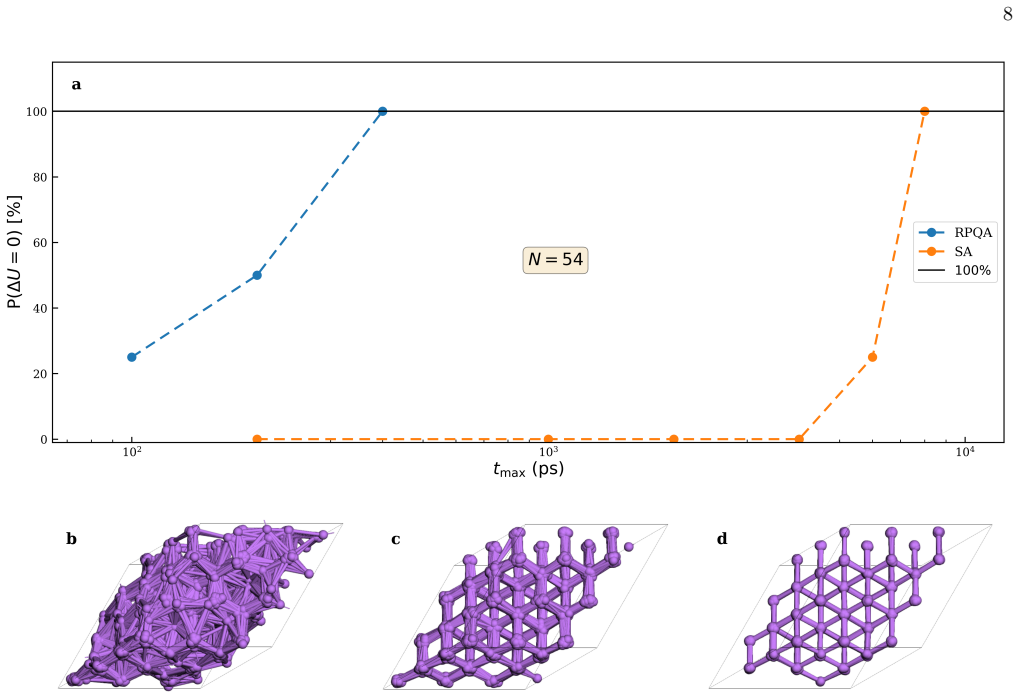
A quantum-annealing protocol based on path-integral molecular dynamics delivers strong performance across atomic systems as either a global optimizer of the potential-energy surface or a quantum-informed structure-search strategy that includes nuclear quantum effects directly in the workflow.
What carries the argument
The path-integral molecular dynamics framework sampling the quantum nuclear density to drive annealing without explicit many-body wavefunction manipulation.
Load-bearing premise
Path-integral molecular dynamics faithfully realizes quantum annealing dynamics without explicit manipulation of many-body wavefunctions.
What would settle it
A benchmark atomic system with a known global minimum where the PIMD-based annealing consistently returns higher-energy configurations than classical simulated annealing or established quantum methods.
Figures
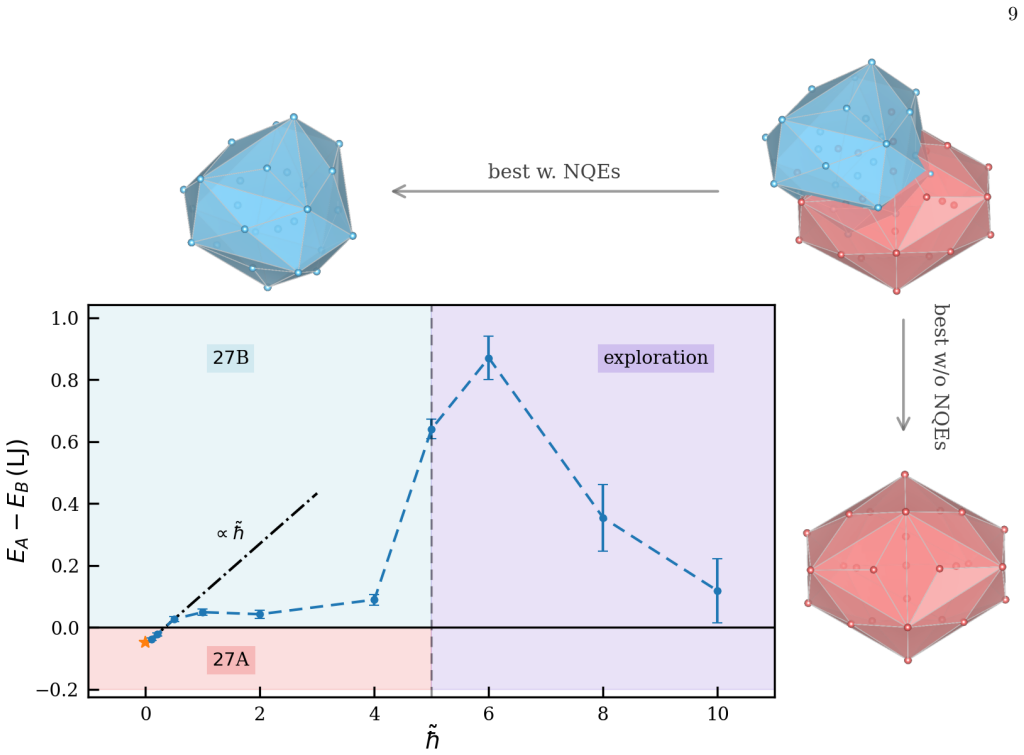
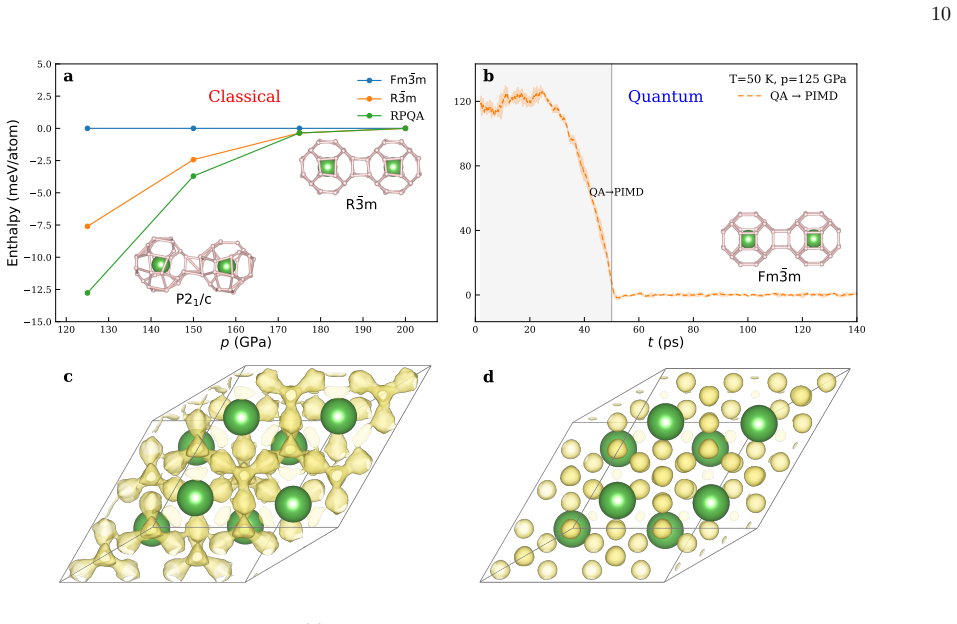
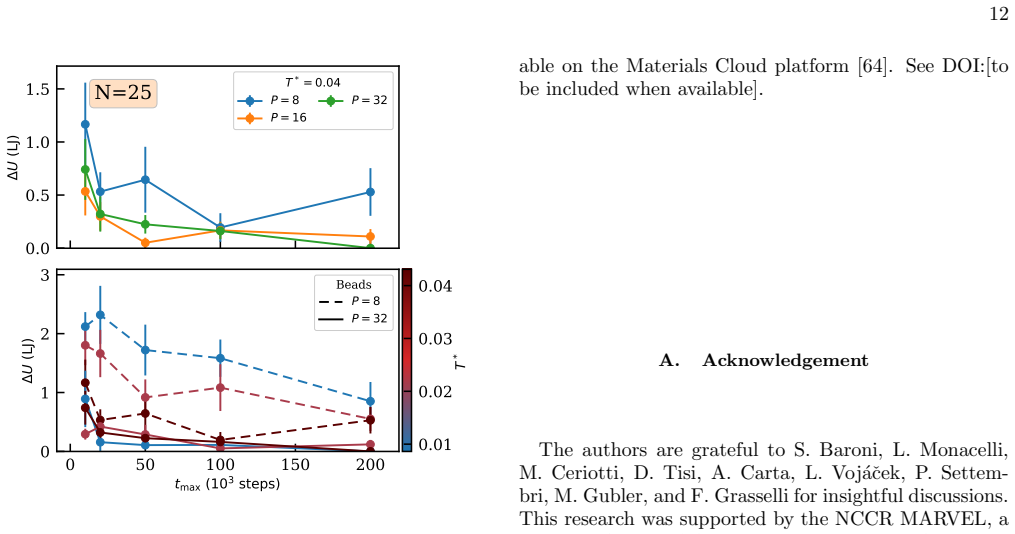
read the original abstract
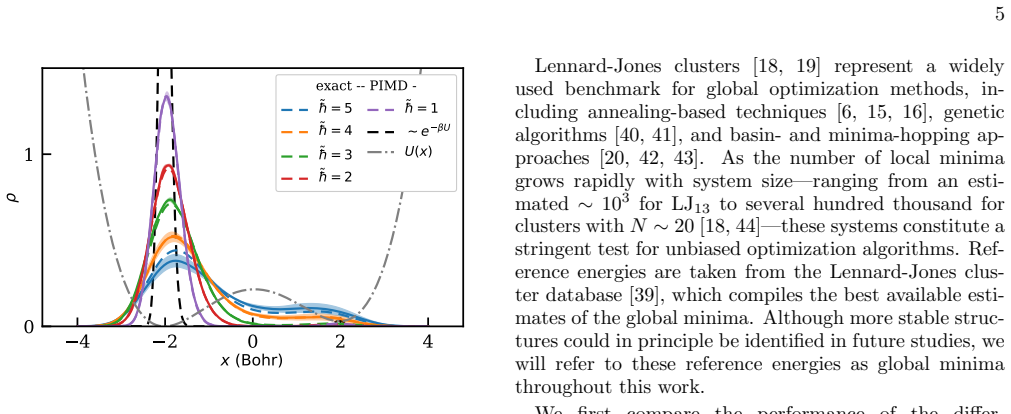
Finding the global minimum of a potential energy surface is a fundamental challenge in materials science, with applications ranging from protein folding to cluster physics and, more broadly, to systems in which the number of (meta)stable configurations grows prohibitively large. In recent decades, quantum annealing (QA) has emerged as a promising global optimization strategy, exploiting quantum fluctuations in contrast to the thermal fluctuations that drive its classical counterpart. Here, we introduce a novel implementation of QA based on path-integral molecular dynamics, an efficient and well-established framework for sampling the quantum nuclear density without the need to manipulate many-body wavefunctions explicitly. While retaining the flexibility and simplicity of molecular dynamics simulations, this quantum-annealing protocol delivers strong performance across a wide range of atomic systems, simulated by either empirical force fields or machine-learning interatomic potentials. The method can be used either as a global optimizer of the potential-energy surface, or as a quantum-informed structure-search strategy in which nuclear quantum effects are included directly in the optimization workflow -- a feature particularly relevant for materials such as high-pressure hydrides.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript introduces a novel quantum annealing (QA) protocol implemented using path-integral molecular dynamics (PIMD) for global minimization of potential energy surfaces. It claims this approach exploits quantum fluctuations for optimization across atomic systems (using empirical force fields or ML interatomic potentials), operates without explicit many-body wavefunction manipulation, and can incorporate nuclear quantum effects directly, with relevance to high-pressure hydrides.
Significance. If the central claim holds and the PIMD schedule is shown to realize controlled QA (distinct from standard PIMD or classical annealing), the method could provide a practical, scalable route to quantum-informed global structure search in materials science. The avoidance of explicit wavefunctions and compatibility with existing MD frameworks would be practical strengths, but no benchmarks, error bars, or convergence data are supplied to assess performance.
major comments (2)
- [Abstract] Abstract: the central claim that the PIMD protocol 'delivers strong performance' via quantum annealing (controlled reduction of quantum fluctuations enabling tunneling-assisted escape) is not supported by any description of the time-dependent schedule, verification against standard PIMD, or numerical evidence; without this, the distinction from ordinary quantum sampling or ergodic exploration is not established.
- [Abstract] Abstract: the assertion that the method works 'without the need to manipulate many-body wavefunctions explicitly' is presented as an advantage, but the mapping from PIMD imaginary-time paths to an annealing schedule (e.g., varying effective ħ or transverse field) is non-trivial and requires explicit demonstration that the dynamics converge via quantum effects rather than thermal sampling; this is load-bearing for the QA label.
minor comments (1)
- [Abstract] The abstract provides no equations, pseudocode, or section references for the schedule implementation, making it impossible to assess reproducibility from the given text.
Simulated Author's Rebuttal
We thank the referee for the careful reading of our manuscript and the constructive comments on the abstract. We address each point below and indicate where revisions will be made to clarify the quantum-annealing protocol and its supporting evidence.
read point-by-point responses
-
Referee: [Abstract] Abstract: the central claim that the PIMD protocol 'delivers strong performance' via quantum annealing (controlled reduction of quantum fluctuations enabling tunneling-assisted escape) is not supported by any description of the time-dependent schedule, verification against standard PIMD, or numerical evidence; without this, the distinction from ordinary quantum sampling or ergodic exploration is not established.
Authors: The full manuscript (Section 2.2 and Figures 3–5) specifies the annealing schedule through a controlled, time-dependent reduction of the effective ħ in the path-integral representation, together with direct comparisons to both classical MD and fixed-ħ PIMD runs on the same systems. These comparisons demonstrate improved escape from local minima attributable to the scheduled quantum fluctuations. We agree that the abstract does not sufficiently point to this evidence and will revise it to reference the schedule description and performance metrics explicitly. revision: yes
-
Referee: [Abstract] Abstract: the assertion that the method works 'without the need to manipulate many-body wavefunctions explicitly' is presented as an advantage, but the mapping from PIMD imaginary-time paths to an annealing schedule (e.g., varying effective ħ or transverse field) is non-trivial and requires explicit demonstration that the dynamics converge via quantum effects rather than thermal sampling; this is load-bearing for the QA label.
Authors: PIMD samples the quantum nuclear density via imaginary-time paths by construction, without ever forming an explicit many-body wavefunction; this is the standard computational advantage of the method. The QA mapping is realized by varying the bead number and the effective ħ(t) according to a prescribed annealing protocol (detailed in Section 2.3), which is shown to produce tunneling-assisted transitions beyond what is obtained at fixed ħ or in the classical limit. We will add a short clarifying paragraph in the revised manuscript that explicitly connects the PIMD parameters to the QA schedule and includes a supplementary comparison isolating the quantum contribution. revision: yes
Circularity Check
No circularity detected; protocol introduced as independent method
full rationale
The paper introduces a novel QA implementation via PIMD without any equations, fitted parameters, or self-citations appearing in the abstract or described claims. No derivation chain is presented that reduces a prediction or result to its inputs by construction, nor are there load-bearing self-citations, ansatzes smuggled via prior work, or renamings of known results. The central description is a methodological proposal whose performance claims are positioned for external validation rather than being tautological or self-referential.
Axiom & Free-Parameter Ledger
Reference graph
Works this paper leans on
-
[1]
W. H. Press, B. P. Flannery, S. A. Teukolsky, and W. T. Vetterling,Numerical recipes in Fortran: diskettes 3.5”(720K) for IBM PC, PS/2 and compatibles (DOS) (Cambridge Univ. Press, 1992)
1992
-
[2]
Kirkpatrick, C
S. Kirkpatrick, C. D. Gelatt Jr, and M. P. Vecchi, Optimization by simulated annealing, Science220, 671 (1983)
1983
-
[3]
Kadowaki and H
T. Kadowaki and H. Nishimori, Quantum annealing in the transverse ising model, Physical Review E58, 5355 (1998)
1998
-
[4]
Brooke, D
J. Brooke, D. Bitko, T. F. Rosenbaum, and G. Aeppli, Quantum annealing of a disordered magnet, Science284, 779 (1999)
1999
-
[5]
E. Farhi, J. Goldstone, S. Gutmann, and M. Sipser, Quantum computation by adiabatic evolution, arXiv preprint quant-ph/0001106 (2000)
Pith/arXiv arXiv 2000
-
[6]
Amara, D
P. Amara, D. Hsu, and J. E. Straub, Global energy mini- mum searches using an approximate solution of the imag- inary time schr¨ odinger equation, The Journal of Physical Chemistry97, 6715 (1993)
1993
-
[7]
G. E. Santoro, R. Marton´ ak, E. Tosatti, and R. Car, The- ory of quantum annealing of an ising spin glass, Science 295, 2427 (2002)
2002
-
[8]
Stella, G
L. Stella, G. E. Santoro, and E. Tosatti, Optimization by quantum annealing: Lessons from simple cases, Physical Review B72, 014303 (2005)
2005
-
[9]
G. E. Santoro and E. Tosatti, Optimization using quan- tum mechanics: quantum annealing through adiabatic evolution, Journal of Physics A: Mathematical and Gen- eral39, R393 (2006)
2006
-
[10]
Stella, G
L. Stella, G. E. Santoro, and E. Tosatti, Monte carlo studies of quantum and classical annealing on a double well, Physical Review B73, 144302 (2006)
2006
-
[11]
G. Lami, P. Torta, G. E. Santoro, and M. Collura, Quan- tum annealing for neural network optimization problems: A new approach via tensor network simulations, SciPost Physics14, 117 (2023)
2023
-
[12]
M. W. Johnson, M. H. Amin, S. Gildert, T. Lanting, F. Hamze, N. Dickson, R. Harris, A. J. Berkley, J. Jo- hansson, P. Bunyk,et al., Quantum annealing with man- ufactured spins, Nature473, 194 (2011)
2011
-
[13]
Perdomo-Ortiz, N
A. Perdomo-Ortiz, N. Dickson, M. Drew-Brook, G. Rose, and A. Aspuru-Guzik, Finding low-energy conformations of lattice protein models by quantum annealing, Scientific Reports2, 571 (2012)
2012
-
[14]
Barends, A
R. Barends, A. Shabani, L. Lamata, J. Kelly, A. Mezza- capo, U. L. Heras, R. Babbush, A. G. Fowler, B. Camp- bell, Y. Chen,et al., Digitized adiabatic quantum com- puting with a superconducting circuit, Nature534, 222 (2016)
2016
-
[15]
A. B. Finnila, M. A. Gomez, C. Sebenik, C. Stenson, and J. D. Doll, Quantum annealing: A new method for min- imizing multidimensional functions, Chemical Physics Letters219, 343 (1994)
1994
-
[16]
Gregor and R
T. Gregor and R. Car, Minimization of the potential en- ergy surface of lennard–jones clusters by quantum opti- mization, Chemical physics letters412, 125 (2005)
2005
-
[17]
Inack and S
E. Inack and S. Pilati, Simulated quantum annealing of double-well and multiwell potentials, Physical Review E 92, 053304 (2015). 13
2015
-
[18]
M. R. Hoare and J. McInnes, Statistical mechanics and morphology of very small atomic clusters, Faraday Dis- cussions of the Chemical Society61, 12 (1976)
1976
-
[19]
J. A. Northby, Structure and binding of lennard-jones clusters: 13≤n≤147, The Journal of Chemical Physics 87, 6166 (1987)
1987
-
[20]
D. J. Wales and J. P. Doye, Global optimization by basin- hopping and the lowest energy structures of lennard-jones clusters containing up to 110 atoms, The Journal of Phys- ical Chemistry A101, 5111 (1997)
1997
-
[21]
Drozdov, P
A. Drozdov, P. Kong, V. Minkov, S. Besedin, M. Ku- zovnikov, S. Mozaffari, L. Balicas, F. F. Balakirev, D. Graf, V. Prakapenka,et al., Superconductivity at 250 k in lanthanum hydride under high pressures, Nature 569, 528 (2019)
2019
-
[22]
Errea, M
I. Errea, M. Calandra, C. J. Pickard, J. R. Nelson, R. J. Needs, Y. Li, H. Liu, Y. Zhang, Y. Ma, and F. Mauri, Quantum hydrogen-bond symmetrization in the super- conducting hydrogen sulfide system, Nature532, 81 (2016)
2016
-
[23]
Monacelli, M
L. Monacelli, M. Casula, K. Nakano, S. Sorella, and F. Mauri, Quantum phase diagram of high-pressure hy- drogen, Nature Physics19, 845 (2023)
2023
-
[24]
Errea, F
I. Errea, F. Belli, L. Monacelli, A. Sanna, T. Koretsune, T. Tadano, R. Bianco, M. Calandra, R. Arita, F. Mauri, et al., Quantum crystal structure in the 250-kelvin super- conducting lanthanum hydride, Nature578, 66 (2020)
2020
-
[25]
D. Poletaev and A. Oganov, Sscha-based evo- lutionary crystal structure prediction at finite temperatures with account for quantum nu- clear motion, arXiv preprint arXiv:2512.24849 https://doi.org/10.48550/arXiv.2512.24849 (2025)
-
[26]
L. E. Smith, P. Settembri, A. Cucciari, L. Boeri, G. Profeta, and S. Hammes-Schiffer, Capturing nuclear quantum effects in high-pressure superconducting hy- drides and ice with nuclear–electronic orbital theory, Proceedings of the National Academy of Sciences123, e2605545123 (2026)
2026
-
[27]
Calvo, J
F. Calvo, J. Doye, and D. Wales, Quantum partition functions from classical distributions: Application to rare-gas clusters, The Journal of Chemical Physics114, 7312 (2001)
2001
-
[28]
Born and V
M. Born and V. Fock, Beweis des adiabatensatzes, Zeitschrift f¨ ur Physik51, 165 (1928)
1928
-
[29]
R. P. Feynman, A. R. Hibbs, and G. H. Weiss, Quantum mechanics and path integrals (1966)
1966
-
[30]
Herman, E
M. Herman, E. Bruskin, and B. Berne, On path inte- gral monte carlo simulations, The Journal of Chemical Physics76, 5150 (1982)
1982
-
[31]
Militzer and D
B. Militzer and D. Ceperley, Path integral monte carlo simulation of the low-density hydrogen plasma, Physical Review E63, 066404 (2001)
2001
-
[32]
Ceriotti, J
M. Ceriotti, J. More, and D. E. Manolopoulos, i-pi: A python interface for ab initio path integral molecular dy- namics simulations, Computer Physics Communications 185, 1019 (2014)
2014
-
[33]
Kapil, M
V. Kapil, M. Rossi, O. Marsalek, R. Petraglia, Y. Lit- man, T. Spura, B. Cheng, A. Cuzzocrea, R. H. Meißner, D. M. Wilkins,et al., i-pi 2.0: A universal force engine for advanced molecular simulations, Computer Physics Communications236, 214 (2019)
2019
-
[34]
Y. Litman, V. Kapil, Y. M. Feldman, D. Tisi, T. Beguˇ si´ c, K. Fidanyan, G. Fraux, J. Higer, M. Kellner, T. E. Li, et al., i-pi 3.0: A flexible and efficient framework for ad- vanced atomistic simulations, The Journal of Chemical Physics161, 10.1063/5.0215869 (2024)
-
[35]
A. P´ erez, M. E. Tuckerman, and M. H. M¨ user, A com- parative study of the centroid and ring-polymer molecu- lar dynamics methods for approximating quantum time correlation functions from path integrals, The Journal of Chemical Physics130, 10.1063/1.3126950 (2009)
-
[36]
Hirshberg, V
B. Hirshberg, V. Rizzi, and M. Parrinello, Path integral molecular dynamics for bosons, Proceedings of the Na- tional Academy of Sciences116, 21445 (2019)
2019
-
[37]
Y. M. Feldman and B. Hirshberg, Quadratic scaling bosonic path integral molecular dynamics, The Journal of Chemical Physics159, 10.1063/5.0173749 (2023)
-
[38]
I. R. Craig and D. E. Manolopoulos, Chemical reaction rates from ring polymer molecular dynamics, The Journal of chemical physics122(2005)
2005
-
[39]
D. J. Wales, J. P. K. Doye, A. Dullweber, M. P. Hodges, F. Y. Naumkin, F. Calvo, J. Hern´ andez-Rojas, and T. F. Middleton, The cambridge cluster database,http: //www-wales.ch.cam.ac.uk/CCD.html
-
[40]
Barr´ on, S
C. Barr´ on, S. G´ omez, D. Romero, and A. Saavedra, A genetic algorithm for lennard-jones atomic clusters, Ap- plied Mathematics Letters12, 85 (1999)
1999
-
[41]
Romero, C
D. Romero, C. Barr´ on, and S. G´ omez, The optimal ge- ometry of lennard-jones clusters: 148–309, Computer Physics Communications123, 87 (1999)
1999
-
[42]
S. Goedecker, Minima hopping: An efficient search method for the global minimum of the potential energy surface of complex molecular systems, The Journal of chemical physics120, 9911 (2004)
2004
-
[43]
Krummenacher, M
M. Krummenacher, M. Gubler, J. A. Finkler, H. Huber, M. Sommer-J¨ orgensen, and S. Goedecker, Performing highly efficient minima hopping structure predictions us- ing the atomic simulation environment (ase), SoftwareX 25, 101632 (2024)
2024
-
[44]
Kostrowicki, L
J. Kostrowicki, L. Piela, B. J. Cherayil, and H. A. Scher- aga, Performance of the diffusion equation method in searches for optimum structures of clusters of lennard- jones atoms, The Journal of Physical Chemistry95, 4113 (1991)
1991
-
[45]
Kennedy and R
J. Kennedy and R. Eberhart, Particle swarm optimiza- tion, inProceedings of ICNN’95 - International Confer- ence on Neural Networks, Vol. 4 (1995) pp. 1942–1948 vol.4
1995
-
[46]
M. R. Bonyadi and Z. Michalewicz, Particle swarm opti- mization for single objective continuous space problems: a review, Evolutionary computation25, 1 (2017)
2017
-
[47]
J. P. Doye, M. A. Miller, and D. J. Wales, Evolution of the potential energy surface with size for lennard-jones clusters, The Journal of Chemical Physics111, 8417 (1999)
1999
-
[48]
Machida, H
A. Machida, H. Saitoh, H. Sugimoto, T. Hattori, A. Sano- Furukawa, N. Endo, Y. Katayama, R. Iizuka, T. Sato, M. Matsuo,et al., Site occupancy of interstitial deu- terium atoms in face-centred cubic iron, Nature Com- munications5, 5063 (2014)
2014
-
[49]
Woi´ nska, S
M. Woi´ nska, S. Grabowsky, P. M. Dominiak, K. Wo´ zniak, and D. Jayatilaka, Hydrogen atoms can be located ac- curately and precisely by x-ray crystallography, Science advances2, e1600192 (2016)
2016
-
[50]
S. P. Huber, M. Minotakis, M. Bercx, T. Reents, K. Eimre, N. Paulish, N. H¨ ormann, M. Uhrin, N. Marzari, and G. Pizzi, Mc3d: The materials cloud computational database of experimentally known stoi- 14 chiometric inorganics, arXiv preprint arXiv:2508.19223 (2025)
arXiv 2025
-
[51]
T. Reents, A. Cantarella, M. Bercx, P. Bonf` a, and G. Pizzi, Score-based diffusion models for accurate crystal-structure inpainting and reconstruction of hydro- gen positions, arXiv preprint arXiv:2601.01959 (2026)
Pith/arXiv arXiv 2026
-
[52]
Z. Fan, Y. Wang, P. Ying, K. Song, J. Wang, Y. Wang, Z. Zeng, K. Xu, E. Lindgren, J. M. Rahm,et al., Gpumd: A package for constructing accurate machine- learned potentials and performing highly efficient atom- istic simulations, The Journal of Chemical Physics157, https://doi.org/10.1063/5.0106617 (2022)
-
[53]
Monacelli, R
L. Monacelli, R. Bianco, M. Cherubini, M. Calandra, I. Errea, and F. Mauri, The stochastic self-consistent har- monic approximation: calculating vibrational properties of materials with full quantum and anharmonic effects, Journal of Physics: Condensed Matter33, 363001 (2021)
2021
-
[54]
Batatia, P
I. Batatia, P. Benner, Y. Chiang, A. M. Elena, D. P. Kov´ acs, J. Riebesell, X. R. Advincula, M. Asta, M. Avay- lon, W. J. Baldwin,et al., A foundation model for atom- istic materials chemistry, The Journal of chemical physics 163(2025)
2025
-
[55]
R. Quhe, M. Nava, P. Tiwary, and M. Parrinello, Path integral metadynamics, Journal of chemical theory and computation11, 1383 (2015)
2015
-
[56]
A. H. Larsen, J. J. Mortensen, J. Blomqvist, I. E. Castelli, R. Christensen, M. Du lak, J. Friis, M. N. Groves, B. Hammer, C. Hargus, E. D. Hermes, P. C. Jennings, P. B. Jensen, J. Kermode, J. R. Kitchin, E. L. Kolsbjerg, J. Kubal, K. Kaasbjerg, S. Lysgaard, J. B. Maronsson, T. Maxson, T. Olsen, L. Pastewka, A. Pe- terson, C. Rostgaard, J. Schiøtz, O. Sch...
2017
-
[57]
Giannozzi, S
P. Giannozzi, S. Baroni, N. Bonini, M. Calandra, R. Car, C. Cavazzoni, D. Ceresoli, G. L. Chiarotti, M. Cococ- cioni, I. Dabo,et al., Quantum espresso: a modular and open-source software project for quantum simula- tions of materials, Journal of physics: Condensed matter 21, 395502 (2009)
2009
-
[58]
Giannozzi, O
P. Giannozzi, O. Andreussi, T. Brumme, O. Bunau, M. Buongiorno Nardelli, M. Calandra, R. Car, C. Cavaz- zoni, D. Ceresoli, M. Cococcioni,et al., Advanced capa- bilities for materials modelling with quantum espresso, Journal of physics: Condensed matter29, 465901 (2017)
2017
-
[59]
Giannozzi, O
P. Giannozzi, O. Baseggio, P. Bonf` a, D. Brunato, R. Car, I. Carnimeo, C. Cavazzoni, S. De Gironcoli, P. Delugas, F. Ferrari Ruffino,et al., Quantum espresso toward the exascale, The Journal of chemical physics152(2020)
2020
-
[60]
S. P. Huber, S. Zoupanos, M. Uhrin, L. Talirz, L. Kahle, R. H¨ auselmann, D. Gresch, T. M¨ uller, A. V. Yakutovich, C. W. Andersen,et al., Aiida 1.0, a scalable computa- tional infrastructure for automated reproducible work- flows and data provenance, Scientific data7, 300 (2020)
2020
-
[61]
Uhrin, S
M. Uhrin, S. P. Huber, J. Yu, N. Marzari, and G. Pizzi, Workflows in aiida: Engineering a high-throughput, event-based engine for robust and modular computa- tional workflows, Computational Materials Science187, 110086 (2021)
2021
-
[62]
Fraux, R
G. Fraux, R. K. Cersonsky, and M. Ceriotti, Chemis- cope: interactive structure-property explorer for materi- als and molecules, Journal of Open Source Software5, 2117 (2020)
2020
-
[63]
Momma and F
K. Momma and F. Izumi, Vesta: a three-dimensional vi- sualization system for electronic and structural analysis, Applied Crystallography41, 653 (2008)
2008
-
[64]
Talirz, S
L. Talirz, S. Kumbhar, E. Passaro, A. V. Yakutovich, V. Granata, F. Gargiulo, M. Borelli, M. Uhrin, S. P. Huber, S. Zoupanos, C. S. Adorf, C. W. Andersen, O. Sch¨ utt, C. A. Pignedoli, D. Passerone, J. VandeVon- dele, T. C. Schulthess, B. Smit, G. Pizzi, and N. Marzari, Materials cloud, a platform for open computational sci- ence, Scientific Data7, 299 (2020)
2020
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.