FMO-xTB: Fragment molecular orbital method with GFN1-xTB for large-scale quantum-mechanical simulations
Pith reviewed 2026-06-29 02:10 UTC · model grok-4.3
The pith
FMO-xTB pairs the fragment molecular orbital method with GFN1-xTB to compute energies and gradients for systems of tens of thousands of atoms with near-linear scaling.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
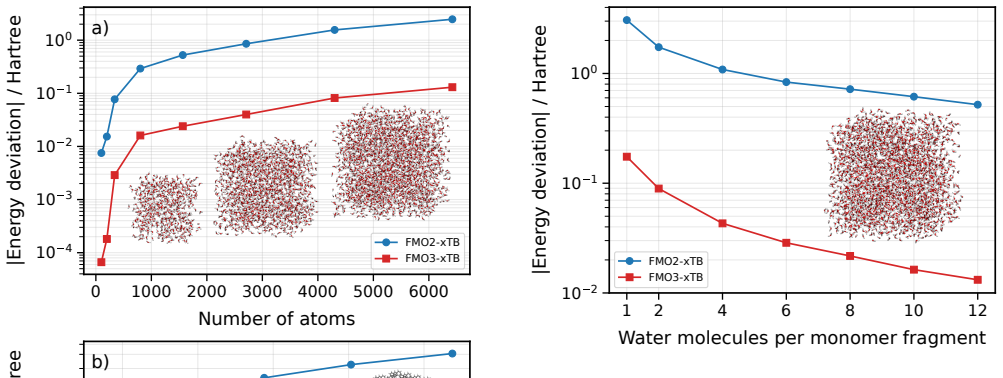
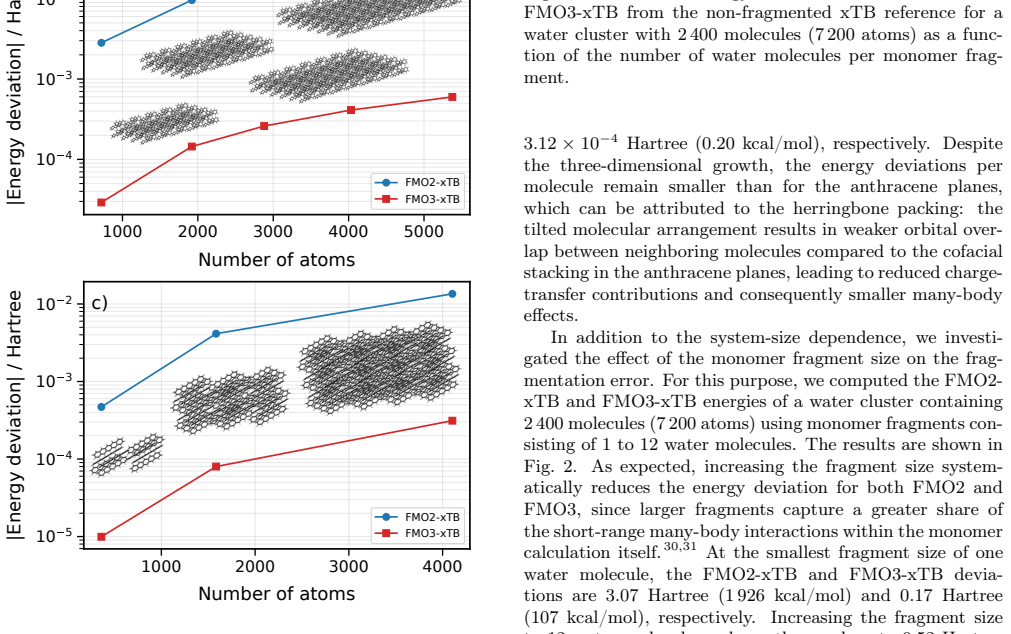
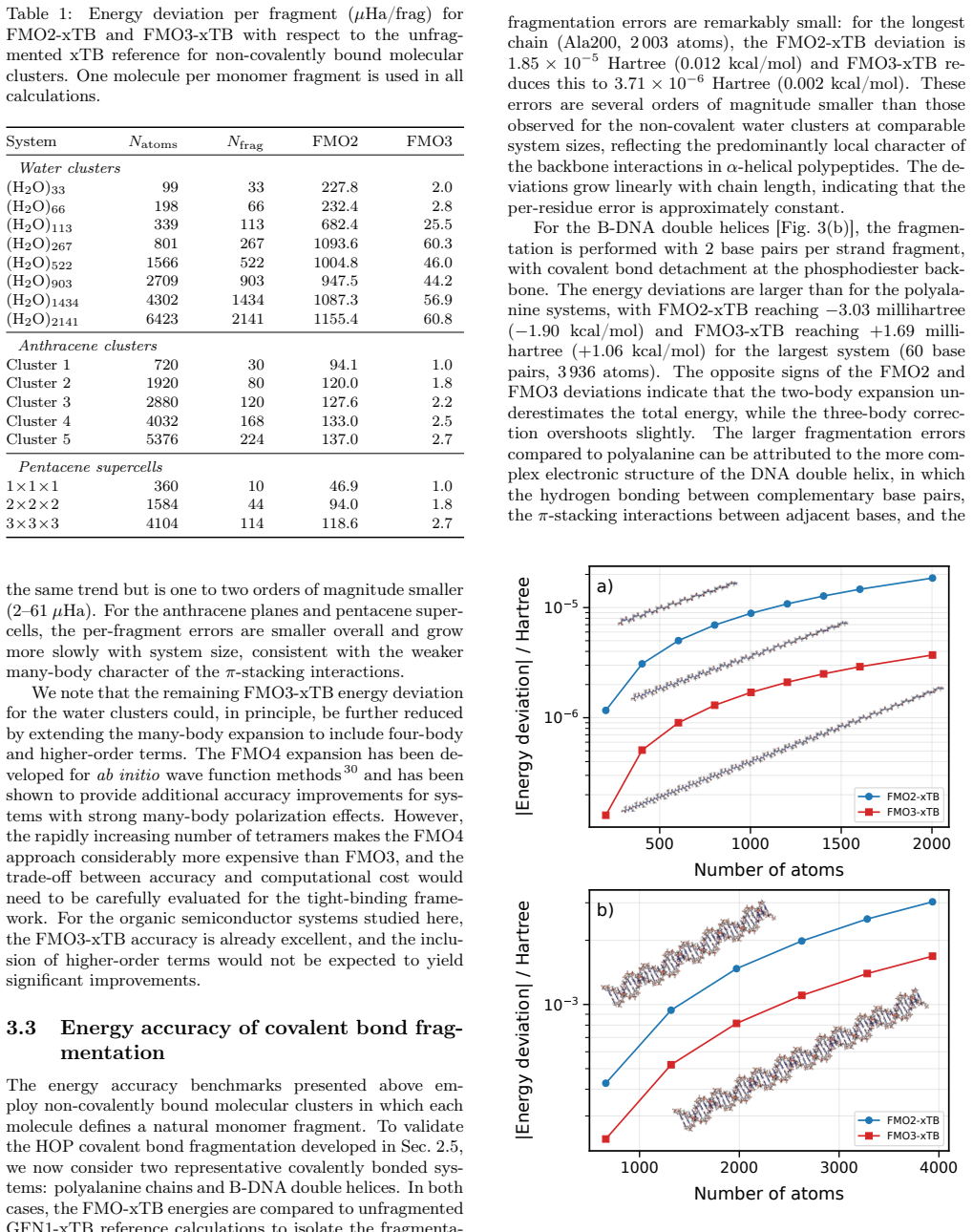
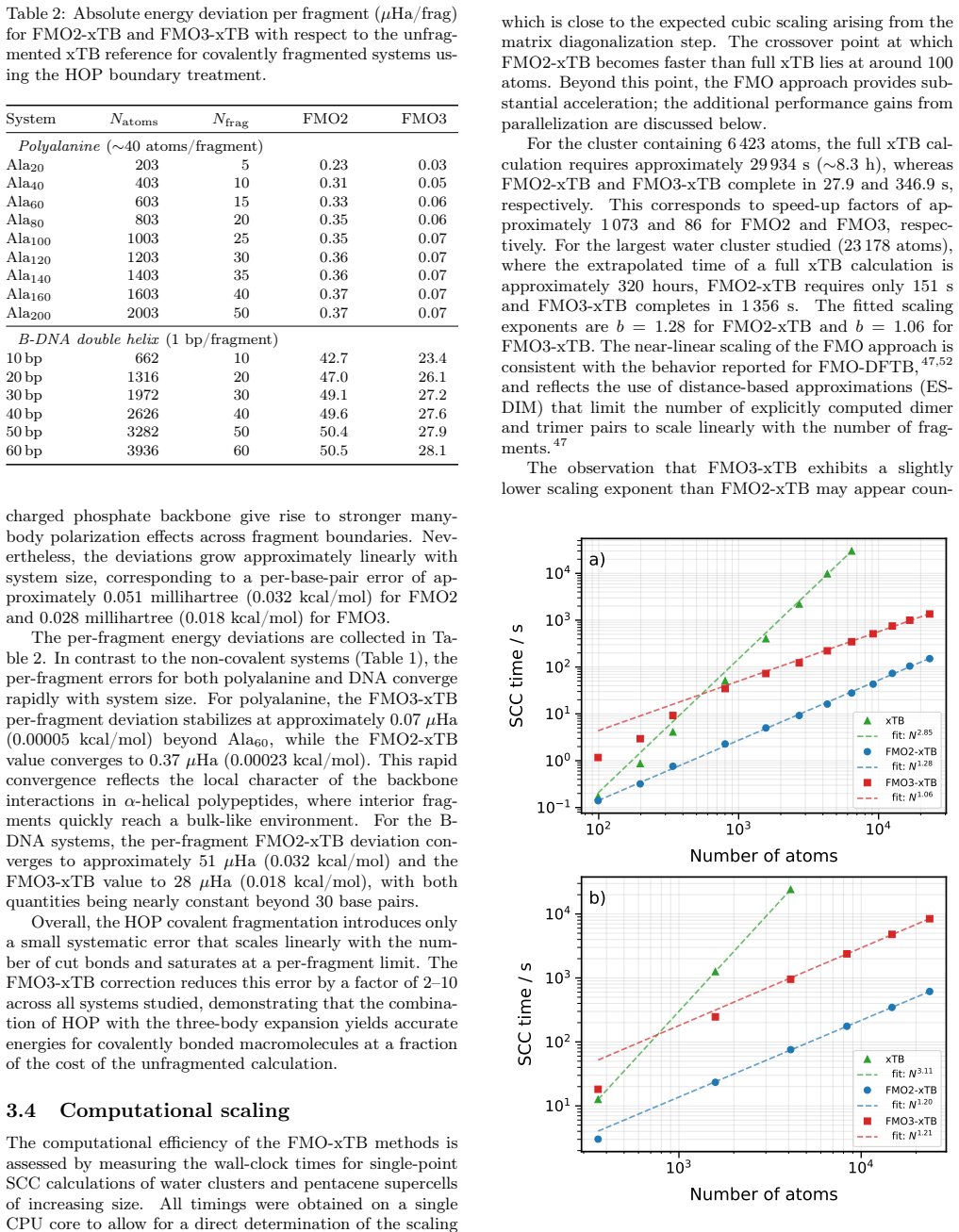
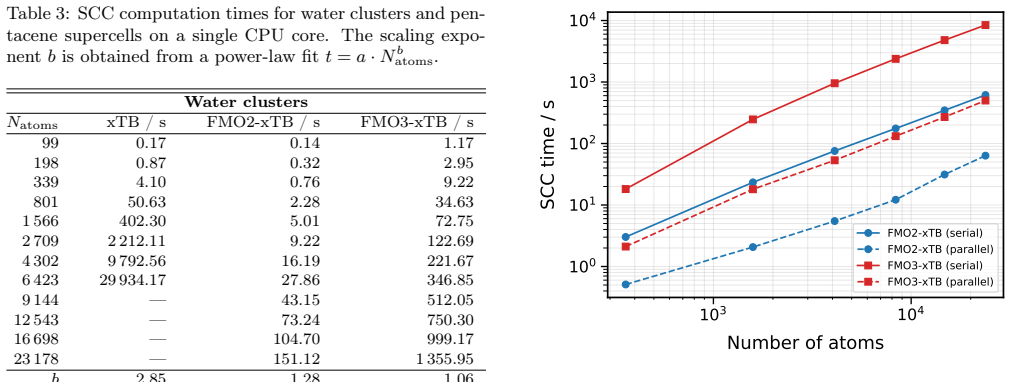
FMO3-xTB reproduces non-fragmented xTB energies within 10^-4 Hartree for organic semiconductor aggregates and within 10^-6 Hartree to millihartree for covalently fragmented polyalanine and B-DNA, while the method as a whole scales with exponents 1.06-1.28 and supports fully analytic energy-plus-gradient evaluations on systems containing more than 20,000 atoms.
What carries the argument
The three-body fragment molecular orbital expansion (FMO3) with GFN1-xTB, using hybrid orbital projection to treat covalent bond fragmentation and self-consistent embedding, supplies the central mechanism that reduces cubic scaling to near-linear while preserving analytic gradients.
If this is right
- Energy and gradient evaluations become feasible for molecular dynamics of organic semiconductor crystals and biomolecular complexes with 20,000+ atoms on single nodes.
- The same framework extends element coverage to all spd-block atoms up to radon without requiring atom-pair-specific parameters.
- Parallel execution over multiple CPU cores further reduces wall time for supercell calculations that previously scaled cubically.
- Routine simulation of systems that were previously accessible only with classical force fields now becomes possible at the quantum-mechanical level.
Where Pith is reading between the lines
- The reported scaling suggests that even larger assemblies, such as protein-ligand complexes or extended material interfaces, could be treated directly rather than through embedding approximations.
- Because gradients are analytic, the method can be dropped into existing molecular-dynamics packages without additional finite-difference overhead.
- The millihartree-level accuracy for B-DNA opens a route to quantum-mechanical sampling of conformational changes in nucleic acids that were previously limited to smaller model systems.
Load-bearing premise
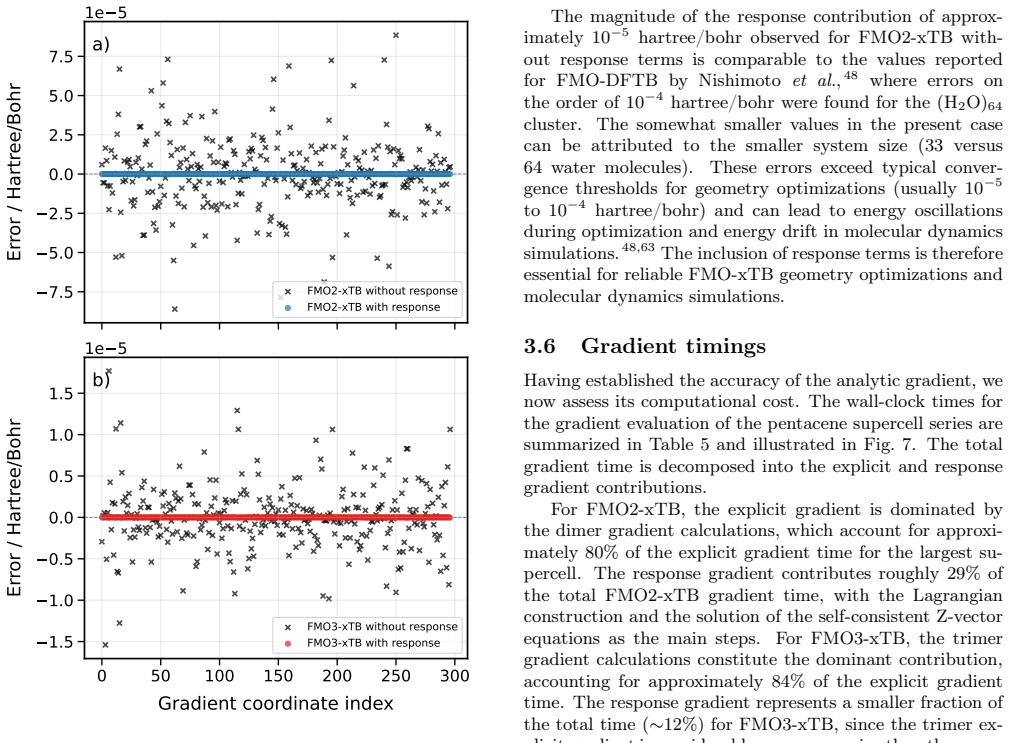
The hybrid orbital projection scheme for cutting covalent bonds adds only errors small enough to stay inside the reported millihartree accuracy window for the tested alpha-helices and DNA structures.
What would settle it
Compute the FMO3-xTB energy and gradient for a covalently bonded system larger than the B-DNA test case and compare directly to a full non-fragmented GFN1-xTB reference; a deviation exceeding a few millihartree would falsify the accuracy claim.
Figures
read the original abstract
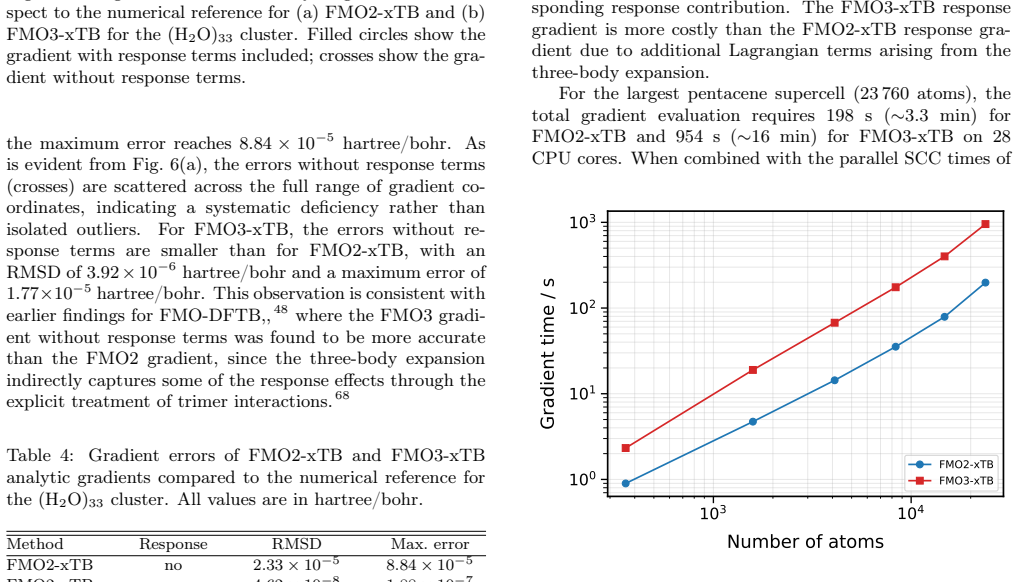
We present the fragment molecular orbital method (FMO) combined with the GFN1-xTB extended tight-binding approach (FMO-xTB) for efficient quantum-mechanical calculations of large molecular systems. Both the two-body (FMO2) and three-body (FMO3) expansions are formulated, and fully analytic energy gradients including the response contribution from the self-consistent embedding potential are derived and implemented. The FMO-xTB method inherits the broad element coverage of GFN1-xTB, which employs element-specific rather than atom-pair-specific parameters and is parameterized for all spd-block elements up to radon(Z = 86), representing a significant practical advantage over FMO- DFTB approaches. The accuracy of FMO-xTB is systematically benchmarked against non-fragmented xTB calculations for water clusters, anthracene aggregates, and pentacene supercells. FMO3-xTB reproduces the reference energies with deviations on the order of 10^-4 Hartree for organic semiconductor systems. The covalent bond fragmentation capability using the hybrid orbital projection (HOP) boundary treatment is also implemented with fully analytic gradients and validated for polyalanine alpha-helices and B-DNA double helices, yielding FMO3-xTB energy deviations on the order of 10^-6 Hartree for polyalanine and in the millihartree range for B-DNA. Near-linear scaling is achieved with effective scaling exponents between b= 1.06 and b= 1.28, compared to cubic scaling for non-fragmented xTB. Parallelization over multiple CPU cores yields significant speed ups, and a complete energy and gradient evaluation of a pentacene supercell containing 23760 atoms is feasible within minutes on a single computing node, enabling routine molecular dynamics simulations of systems with tens of thousands of atoms. The method is implemented in the DIALECT software package.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript introduces the FMO-xTB method, combining the fragment molecular orbital (FMO) approach with GFN1-xTB for efficient QM calculations on large systems. It formulates both FMO2 and FMO3 expansions, derives and implements fully analytic energy gradients (including response terms from the embedding potential), benchmarks FMO3-xTB energies against non-fragmented xTB on water clusters, anthracene aggregates, and pentacene supercells (deviations ~10^{-4} Ha for organic semiconductors), validates the hybrid orbital projection (HOP) for covalent fragmentation on polyalanine alpha-helices (~10^{-6} Ha) and B-DNA (~millihartree), and reports near-linear scaling (exponents 1.06-1.28) with a 23760-atom pentacene example completing in minutes.
Significance. If the accuracy and scaling claims hold, FMO-xTB would provide a practical route to routine QM-level MD on systems of tens of thousands of atoms while retaining GFN1-xTB's broad elemental coverage (all spd elements to Z=86). The independent benchmarks against full xTB calculations and the implementation of analytic gradients are notable strengths that support reproducibility and usability.
major comments (1)
- [Abstract and covalent fragmentation validation] Abstract and covalent fragmentation section: The hybrid orbital projection (HOP) treatment is validated only for polyalanine alpha-helices and B-DNA double helices. No data are provided for other covalent bond types (e.g., C-C single bonds in hydrocarbons, disulfide bonds) or branched topologies, which limits support for the claim of applicability to general large molecular systems requiring covalent fragmentation.
minor comments (2)
- The scaling exponents (b = 1.06 to 1.28) are reported without specifying the exact system sizes or fragmentation schemes used to obtain each value; a table or figure reference would improve clarity.
- The manuscript states that FMO-xTB inherits broad element coverage from GFN1-xTB, but no explicit test systems containing heavy elements (Z > 36) are mentioned in the benchmarks.
Simulated Author's Rebuttal
We thank the referee for the positive assessment of our manuscript and the recommendation for minor revision. We address the single major comment below.
read point-by-point responses
-
Referee: [Abstract and covalent fragmentation validation] Abstract and covalent fragmentation section: The hybrid orbital projection (HOP) treatment is validated only for polyalanine alpha-helices and B-DNA double helices. No data are provided for other covalent bond types (e.g., C-C single bonds in hydrocarbons, disulfide bonds) or branched topologies, which limits support for the claim of applicability to general large molecular systems requiring covalent fragmentation.
Authors: We agree that the HOP validation is restricted to polyalanine α-helices and B-DNA, which primarily feature peptide and phosphodiester linkages. These systems were chosen because they represent the most common use cases for covalent fragmentation in large biomolecules, the primary target application of FMO-xTB. While the HOP procedure itself is a standard, transferable technique from the FMO literature and our implementation follows the established formulation without system-specific adjustments, we acknowledge that explicit benchmarks on additional bond types (e.g., alkane C–C or disulfides) and branched topologies would strengthen the generality claim. Because the current manuscript already demonstrates sub-millihartree accuracy on the tested covalent cases together with fully analytic gradients, we do not believe the limitation undermines the core technical contribution. We are prepared to insert a brief clarifying sentence in the abstract and methods section noting the scope of the HOP benchmarks if the editor considers it necessary. revision: partial
Circularity Check
No significant circularity; central claims rest on independent benchmarks against full xTB
full rationale
The paper formulates FMO2/FMO3-xTB by direct combination of the standard fragment molecular orbital method with the existing GFN1-xTB Hamiltonian, derives analytic gradients from the embedding potential, and reports accuracy via explicit numerical comparison to non-fragmented reference xTB calculations on water clusters, anthracene aggregates, pentacene supercells, polyalanine, and B-DNA. No equation reduces a claimed result to a fitted parameter or self-defined quantity by construction, and no load-bearing premise is justified solely by self-citation. The HOP boundary treatment is presented as an implemented feature whose errors are measured empirically on the cited test systems rather than asserted by definition. This satisfies the default expectation of a non-circular derivation chain.
Axiom & Free-Parameter Ledger
axioms (1)
- domain assumption The total energy can be approximated by a truncated many-body expansion over fragment energies plus interaction corrections.
Reference graph
Works this paper leans on
-
[1]
A non-self-consistent tight-binding electronic structure potential in a polarized double-. J. Chem. Phys. , author =. 2023 , pages =. doi:10.1063/5.0137838 , language =
-
[2]
A. J. Chem. Theory Comput. , author =. 2017 , pages =. doi:10.1021/acs.jctc.7b00118 , number =
-
[3]
Divide-. J. Comput. Chem. , author =. 2025 , keywords =. doi:10.1002/jcc.70255 , abstract =
-
[4]
Fedorov, Dmitri G. and Inostroza, Diego and Courbiere, Bastien and Guegan, Fréderic and Contreras-García, Julia and Mori, Seiji , month = feb, year =. Decomposition. J. Chem. Theory Comput. , publisher =. doi:10.1021/acs.jctc.4c01654 , abstract =
-
[5]
Einsele, Richard and Miao, Xincheng and Philipp, Luca Nils and Mitrić, Roland , month = jul, year =. J. Phys. Chem. A , publisher =. doi:10.1021/acs.jpca.5c03307 , abstract =
-
[6]
Use of caps in the auxiliary basis set formulation of the fragment molecular orbital method , volume =. J. Comput. Chem. , author =. 2024 , pages =. doi:10.1002/jcc.27345 , abstract =
-
[7]
J. Chem. Phys. , author =. 2024 , pages =. doi:10.1063/5.0231191 , abstract =
-
[8]
Einsele, Richard and Mitrić, Roland , month = jul, year =. Nonadiabatic. J. Chem. Theory Comput. , publisher =. doi:10.1021/acs.jctc.4c00539 , abstract =
-
[9]
Plett, Christoph and Katbashev, Abylay and Ehlert, Sebastian and Grimme, Stefan and Bursch, Markus , month = jul, year =. Phys. Chem. Chem. Phys. , publisher =. doi:10.1039/D3CP02178E , abstract =
-
[10]
Divide-and-. J. Phys. Chem. A , author =. 2023 , pages =. doi:10.1021/acs.jpca.2c06965 , abstract =
-
[11]
Long-range corrected fragment molecular orbital density functional tight-binding method for excited states in large molecular systems , volume =. J. Chem. Phys. , author =. 2023 , pages =. doi:10.1063/5.0136844 , abstract =
-
[12]
Fragment molecular orbital calculations for biomolecules , volume =. Curr. Opin. Struct. Biol. , author =. 2022 , keywords =. doi:10.1016/j.sbi.2021.08.010 , abstract =
-
[13]
Free. J. Phys. Chem. Lett. , author =. 2022 , pages =. doi:10.1021/acs.jpclett.2c00040 , abstract =
-
[14]
Polarization energies in the fragment molecular orbital method , volume =. J. Comput. Chem. , author =. 2022 , pages =. doi:10.1002/jcc.26869 , abstract =
-
[15]
The fragment molecular orbital method combined with density-functional tight-binding and periodic boundary conditions , volume =. J. Chem. Phys. , author =. 2021 , pages =. doi:10.1063/5.0039520 , abstract =
-
[16]
Analytic first and second derivatives of the energy in the fragment molecular orbital method combined with molecular mechanics , volume =. Int. J. Quantum Chem. , author =. doi:10.1002/qua.26414 , abstract =
-
[17]
The. J. Chem. Theory Comput. , author =. 2019 , pages =. doi:10.1021/acs.jctc.9b00108 , abstract =
-
[18]
Dcdftbmd:. J. Comput. Chem. , author =. 2019 , pages =. doi:10.1002/jcc.25804 , language =
-
[19]
Adaptive frozen orbital treatment for the fragment molecular orbital method combined with density-functional tight-binding , volume =. J. Chem. Phys. , author =. 2018 , pages =. doi:10.1063/1.5012935 , language =
-
[20]
Analytic second derivatives for the efficient electrostatic embedding in the fragment molecular orbital method , volume =. J. Comput. Chem. , author =. 2018 , pages =. doi:10.1002/jcc.25360 , language =
-
[21]
Computational. Angew. Chem. Int. Ed. , author =. 2018 , keywords =. doi:10.1002/anie.201709943 , abstract =
-
[22]
Three-body expansion of the fragment molecular orbital method combined with density-functional tight-binding , volume =. J. Comput. Chem. , author =. 2017 , pages =. doi:10.1002/jcc.24693 , language =
-
[23]
Houk, K. N. and Liu, Fang , month = mar, year =. Holy. Acc. Chem. Res. , publisher =. doi:10.1021/acs.accounts.6b00532 , abstract =
-
[24]
The fragment molecular orbital method combined with density-functional tight-binding and the polarizable continuum model , volume =. Phys. Chem. Chem. Phys. , author =. 2016 , pages =. doi:10.1039/C6CP02186G , abstract =
-
[25]
Analytic second derivative of the energy for density-functional tight-binding combined with the fragment molecular orbital method , volume =. J. Chem. Phys. , author =. 2016 , pages =. doi:10.1063/1.4959231 , language =
-
[26]
Divide-and-. J. Phys. Chem. B , author =. 2016 , pages =. doi:10.1021/acs.jpcb.5b12439 , abstract =
-
[27]
Raghavachari, Krishnan and Saha, Arjun , month = jun, year =. Accurate. Chem. Rev. , publisher =. doi:10.1021/cr500606e , number =
-
[28]
Large-. J. Phys. Chem. Lett. , author =. 2015 , pages =. doi:10.1021/acs.jpclett.5b02490 , abstract =
-
[29]
Third-order density-functional tight-binding combined with the fragment molecular orbital method , volume =. Chem. Phys. Lett. , author =. 2015 , pages =. doi:10.1016/j.cplett.2015.07.022 , language =
-
[30]
Tanaka, Shigenori and Mochizuki, Yuji and Komeiji, Yuto and Okiyama, Yoshio and Fukuzawa, Kaori , month = may, year =. Electron-correlated fragment-molecular-orbital calculations for biomolecular and nano systems , volume =. Phys. Chem. Chem. Phys. , publisher =. doi:10.1039/C4CP00316K , abstract =
-
[31]
Density-. J. Chem. Theory Comput. , author =. 2014 , pages =. doi:10.1021/ct500489d , abstract =
-
[32]
Density functional tight binding: values of semi-empirical methods in an ab initio era , volume =
Cui, Qiang and Elstner, Marcus , month = jun, year =. Density functional tight binding: values of semi-empirical methods in an ab initio era , volume =. Phys. Chem. Chem. Phys. , publisher =. doi:10.1039/C4CP00908H , abstract =
-
[33]
Gordon, Mark S. and Fedorov, Dmitri G. and Pruitt, Spencer R. and Slipchenko, Lyudmila V. , month = jan, year =. Fragmentation. Chem. Rev. , publisher =. doi:10.1021/cr200093j , number =
-
[34]
Fully analytic energy gradient in the fragment molecular orbital method , volume =. J. Chem. Phys. , author =. 2011 , pages =. doi:10.1063/1.3568010 , language =
-
[35]
Gaus, Michael and Cui, Qiang and Elstner, Marcus , month = apr, year =. J. Chem. Theory Comput. , publisher =. doi:10.1021/ct100684s , abstract =
-
[36]
and Kitaura, Kazuo , month = dec, year =
Sawada, Toshihiko and Fedorov, Dmitri G. and Kitaura, Kazuo , month = dec, year =. Binding of. J. Phys. Chem. B , publisher =. doi:10.1021/jp1068895 , abstract =
-
[37]
Importance of the hybrid orbital operator derivative term for the energy gradient in the fragment molecular orbital method , volume =. Chem. Phys. Lett. , author =. 2010 , pages =. doi:10.1016/j.cplett.2010.04.043 , abstract =
-
[38]
Divide-and-conquer self-consistent field calculation for open-shell systems:. Chem. Phys. Lett. , author =. 2010 , pages =. doi:10.1016/j.cplett.2010.10.005 , abstract =
-
[39]
Fedorov, Dmitri G. and Slipchenko, Lyudmila V. and Kitaura, Kazuo , month = aug, year =. Systematic. J. Phys. Chem. A , publisher =. doi:10.1021/jp101724p , abstract =
-
[40]
Angew. Chem. Int. Ed. , author =. 2009 , keywords =. doi:10.1002/anie.200802019 , abstract =
-
[41]
J. Comput. Chem. , author =. 2009 , keywords =. doi:10.1002/jcc.21224 , abstract =
-
[42]
Extension of linear-scaling divide-and-conquer-based correlation method to coupled cluster theory with singles and doubles excitations , volume =. J. Chem. Phys. , author =. 2008 , pages =. doi:10.1063/1.2956490 , language =
-
[43]
Fedorov, Dmitri G. and Jensen, Jan H. and Deka, Ramesh C. and Kitaura, Kazuo , month = nov, year =. Covalent. J. Phys. Chem. A , publisher =. doi:10.1021/jp805435n , abstract =
-
[44]
Extending the. J. Phys. Chem. A , author =. 2007 , pages =. doi:10.1021/jp0716740 , language =
-
[45]
Time-dependent density functional theory based upon the fragment molecular orbital method , volume =. J. Chem. Phys. , author =. 2007 , pages =. doi:10.1063/1.2772850 , language =
-
[46]
Implementation of divide-and-conquer method including. J. Comput. Chem. , author =. 2007 , pages =. doi:10.1002/jcc.20707 , abstract =
-
[47]
The three-body fragment molecular orbital method for accurate calculations of large systems , volume =. Chem. Phys. Lett. , author =. 2006 , pages =. doi:10.1016/j.cplett.2006.10.052 , abstract =
-
[48]
Multilayer. J. Phys. Chem. A , author =. 2005 , pages =. doi:10.1021/jp047186z , language =
-
[49]
The importance of three-body terms in the fragment molecular orbital method , volume =. J. Chem. Phys. , author =. 2004 , pages =. doi:10.1063/1.1687334 , abstract =
-
[50]
Second order. J. Chem. Phys. , author =. 2004 , pages =. doi:10.1063/1.1769362 , abstract =
-
[51]
Fragment molecular orbital method: use of approximate electrostatic potential , volume =. Chem. Phys. Lett. , author =. 2002 , pages =. doi:10.1016/S0009-2614(01)01416-6 , abstract =
-
[52]
Orthogonalization corrections for semiempirical methods , volume =. Theor. Chem. Acc. , author =. 2000 , keywords =. doi:10.1007/s002149900083 , abstract =
-
[53]
Fragment molecular orbital method: application to polypeptides , volume =. Chem. Phys. Lett. , author =. 2000 , pages =. doi:10.1016/S0009-2614(00)00070-1 , abstract =
-
[54]
Fragment molecular orbital method: an approximate computational method for large molecules , volume =. Chem. Phys. Lett. , author =. 1999 , pages =. doi:10.1016/S0009-2614(99)00874-X , abstract =
-
[55]
Journal of Molecular Structure: THEOCHEM , author =
A new. Journal of Molecular Structure: THEOCHEM , author =. 1999 , keywords =. doi:10.1016/S0166-1280(98)00475-8 , abstract =
-
[56]
A density‐matrix divide‐and‐conquer approach for electronic structure calculations of large molecules , volume =. J. Chem. Phys. , author =. 1995 , pages =. doi:10.1063/1.470549 , abstract =
-
[57]
Direct calculation of electron density in density-functional theory , volume =
Yang, Weitao , month = mar, year =. Direct calculation of electron density in density-functional theory , volume =. Phys. Rev. Lett. , publisher =. doi:10.1103/PhysRevLett.66.1438 , abstract =
-
[58]
Optimization of parameters for semiempirical methods. J. Comput. Chem. , author =. 1989 , pages =. doi:10.1002/jcc.540100208 , abstract =
-
[59]
Dewar, Michael J. S. and Zoebisch, Eve G. and Healy, Eamonn F. and Stewart, James J. P. , month = jun, year =. Development and use of quantum mechanical molecular models. 76. J. Am. Chem. Soc. , publisher =. doi:10.1021/ja00299a024 , number =
-
[60]
Dewar, Michael J. S. and Thiel, Walter , month = jun, year =. Ground states of molecules. 38. J. Am. Chem. Soc. , publisher =. doi:10.1021/ja00457a004 , number =
-
[61]
Theoretical studies of enzymic reactions:. J. Mol. Biol. , author =. 1976 , pages =. doi:10.1016/0022-2836(76)90311-9 , abstract =
-
[62]
Approximate. J. Chem. Phys. , author =. 1965 , pages =. doi:10.1063/1.1701475 , abstract =
-
[63]
On the. Chem. Eur. J. , author =. 1999 , keywords =. doi:10.1002/(SICI)1521-3765(19991105)5:11<3399::AID-CHEM3399>3.0.CO;2-V , abstract =
-
[64]
The crystallography of anthracene at 95°. Acta Cryst , author =. 1964 , pages =. doi:10.1107/S0365110X64001281 , abstract =
-
[65]
Mercury 4.0: from visualization to analysis, design and prediction , volume =. J. Appl. Crystallogr. , author =. 2020 , keywords =. doi:10.1107/S1600576719014092 , abstract =
-
[66]
Froitzheim, Thomas and Müller, Marcel and Hansen, Andreas and Grimme, Stefan , month = jun, year =. g-. doi:10.26434/chemrxiv-2025-bjxvt , abstract =
-
[67]
A consistent and accurate. J. Chem. Phys. , author =. 2010 , pages =. doi:10.1063/1.3382344 , number =
-
[68]
Subsystem density-functional theory:. Wiley Interdiscip. Rev. Comput. Mol. Sci. , author =. 2014 , pages =. doi:10.1002/wcms.1175 , number =
-
[69]
Self-consistent-charge density-functional tight-binding method for simulations of complex materials properties , volume =. Phys. Rev. B , author =. 1998 , pages =. doi:10.1103/PhysRevB.58.7260 , number =
-
[70]
Semiempirical. Chem. Rev. , author =. 2016 , pages =. doi:10.1021/acs.chemrev.5b00584 , abstract =
-
[71]
Semiempirical quantum–chemical methods , volume =. Wiley Interdiscip. Rev. Comput. Mol. Sci. , author =. 2014 , pages =. doi:10.1002/wcms.1161 , number =
-
[72]
Development of. J. Chem. Theory Comput. , author =. 2019 , pages =. doi:10.1021/acs.jctc.8b01214 , abstract =
-
[73]
Three pillars for achieving quantum mechanical molecular dynamics simulations of huge systems:. J. Comput. Chem. , author =. 2016 , pages =. doi:10.1002/jcc.24419 , number =
-
[74]
Extended tight‐binding quantum chemistry methods , volume =. Wiley Interdiscip. Rev. Comput. Mol. Sci. , author =. doi:10.1002/wcms.1493 , abstract =
-
[75]
J. Chem. Theory Comput. , author =. 2019 , pages =. doi:10.1021/acs.jctc.8b01176 , abstract =
-
[76]
N.; Liu, F
Houk, K. N.; Liu, F. Holy Grails for Computational Organic Chemistry and Biochemistry . Acc. Chem. Res. 2017, 50, 539--543
2017
-
[77]
Grimme, S.; Schreiner, P. R. Computational Chemistry : The Fate of Current Methods and Future Challenges . Angew. Chem. Int. Ed. 2018, 57, 4170--4176
2018
-
[78]
S.; Fedorov, D
Gordon, M. S.; Fedorov, D. G.; Pruitt, S. R.; Slipchenko, L. V. Fragmentation Methods : A Route to Accurate Calculations on Large Systems . Chem. Rev. 2012, 112, 632--672
2012
-
[79]
Accurate Composite and Fragment - Based Quantum Chemical Models for Large Molecules
Raghavachari, K.; Saha, A. Accurate Composite and Fragment - Based Quantum Chemical Models for Large Molecules . Chem. Rev. 2015, 115, 5643--5677
2015
-
[80]
A.; Santry, D
Pople, J. A.; Santry, D. P.; Segal, G. A. Approximate Self ‐ Consistent Molecular Orbital Theory . I . Invariant Procedures . J. Chem. Phys. 1965, 43, S129--S135
1965
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.