A Combined Tight Binding with Machine Learning Potential Model for Magnesium Compounds
Pith reviewed 2026-06-25 20:09 UTC · model grok-4.3
The pith
Replacing the pair potential in DFTB with a MACE machine learning potential improves accuracy for magnesium systems while retaining electronic structure information.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
The DFTB+MACE model, formed by substituting the repulsive pair potential with a MACE many-body term trained on DFT minus DFTB differences, yields improved accuracy over DFTB plus pair potential for structural, dynamical, and vibrational properties of magnesium compounds while still delivering electronic structures at moderate added computational expense.
What carries the argument
The DFTB+MACE hybrid potential in which MACE replaces the conventional repulsive pair potential and is trained on the DFT-DFTB energy and force residuals.
If this is right
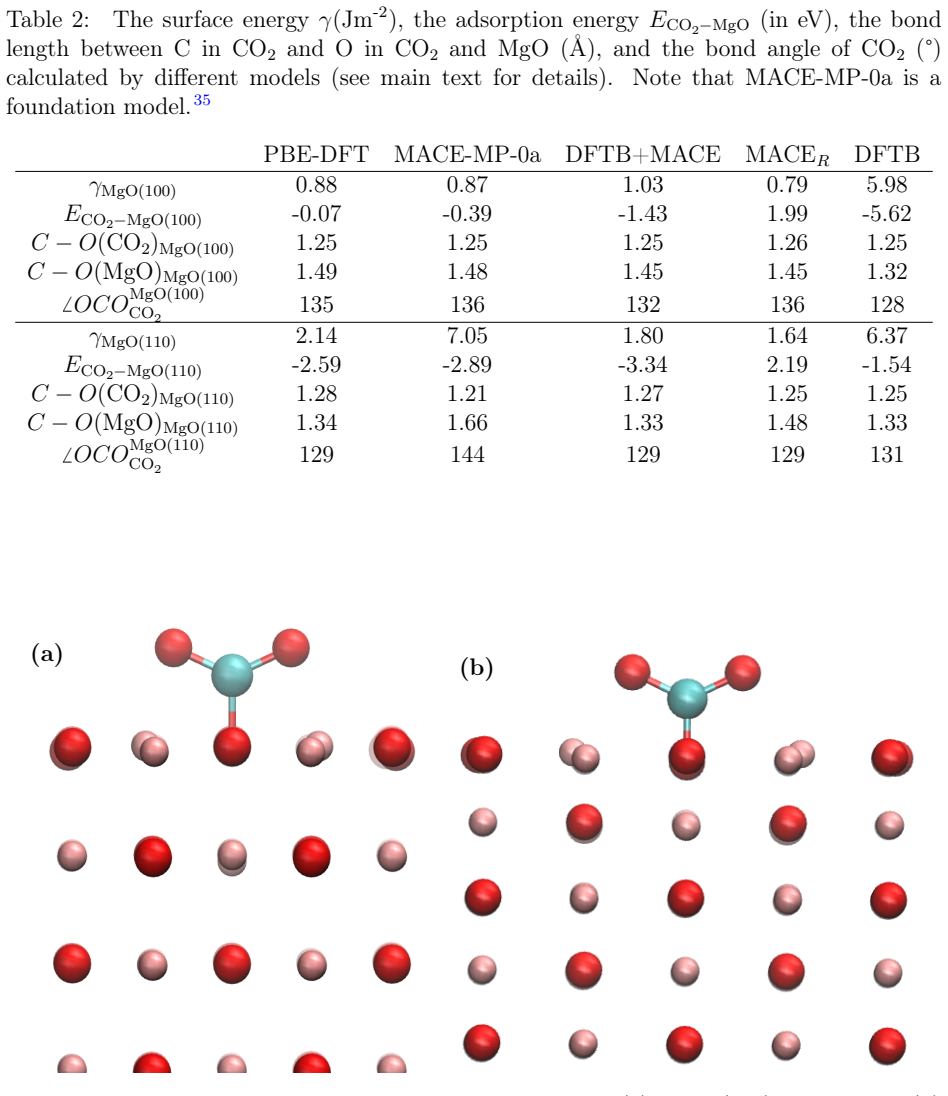
- Structural relaxations of MgO-CO2 adsorption systems reach higher accuracy than with DFTB plus pair potential.
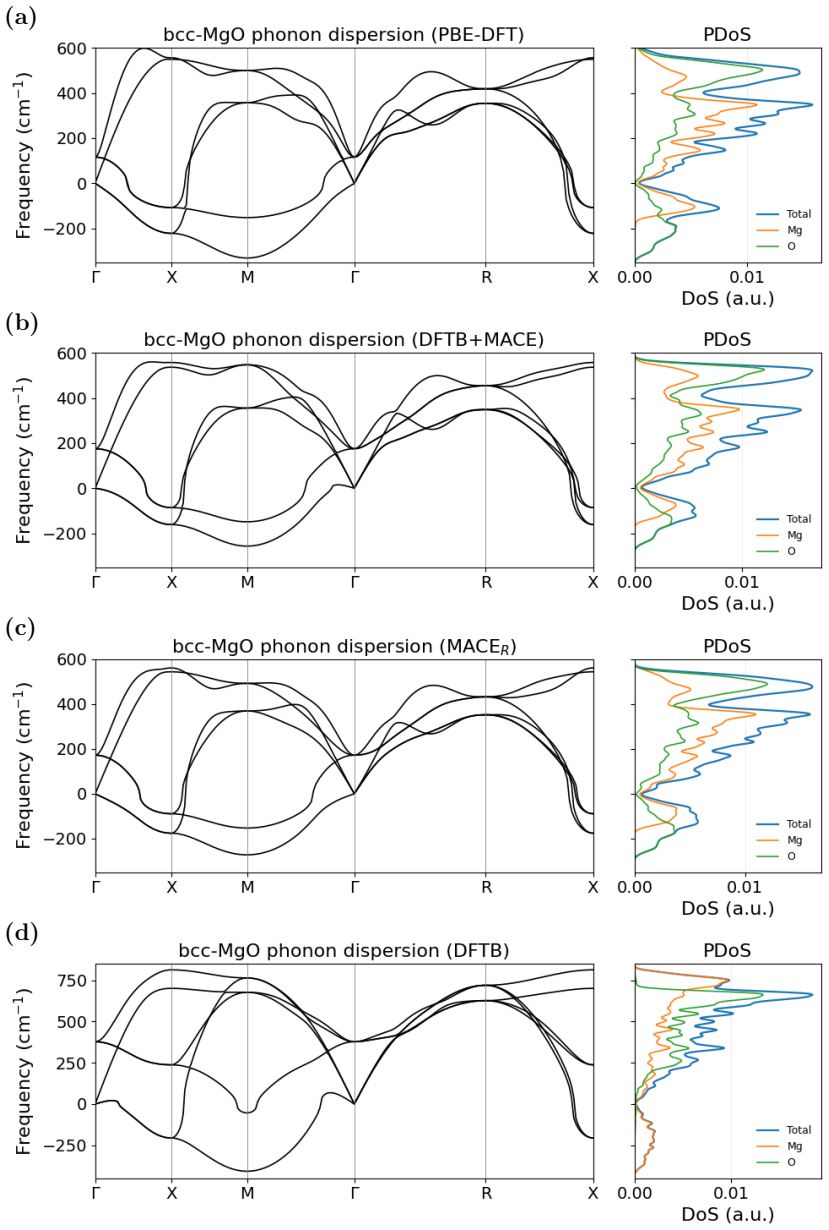
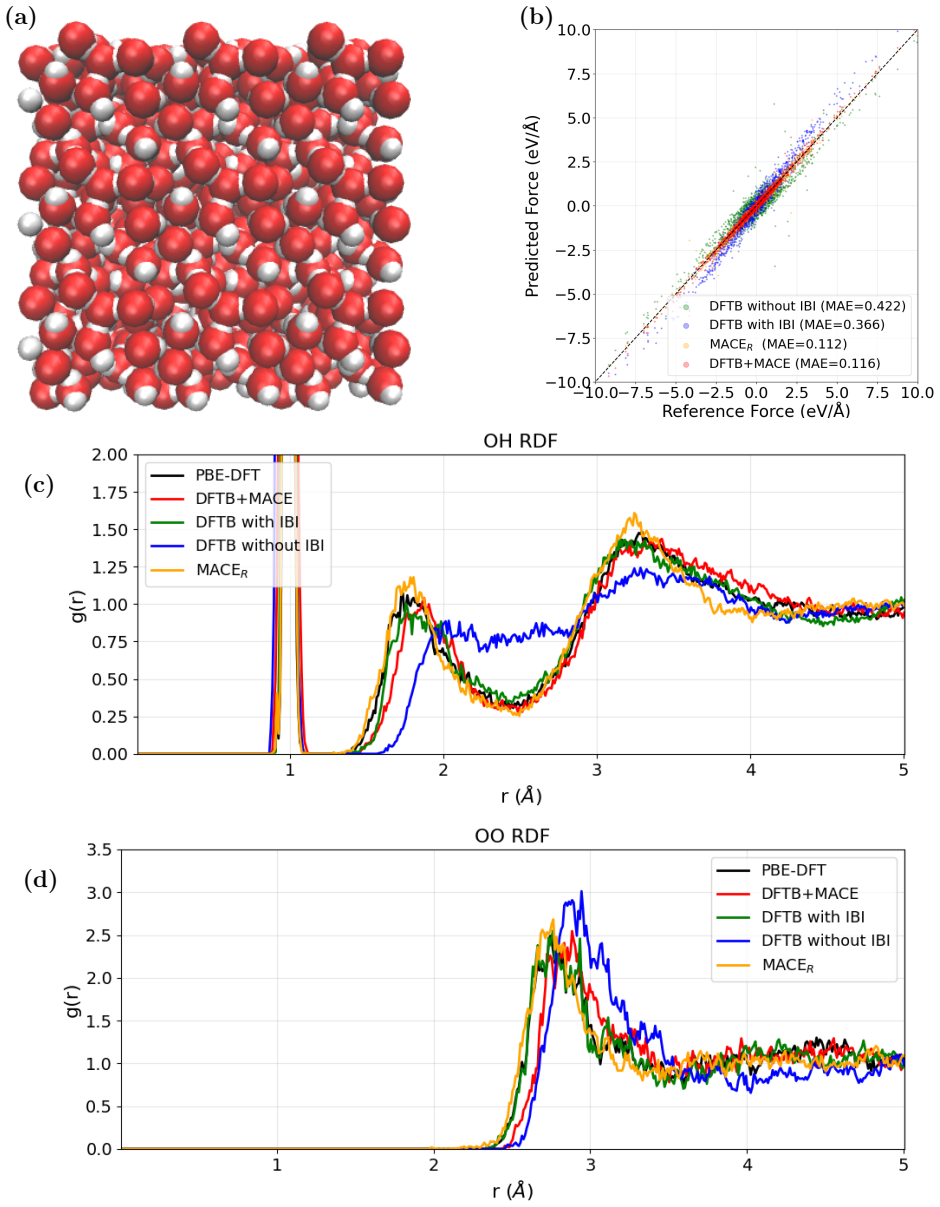
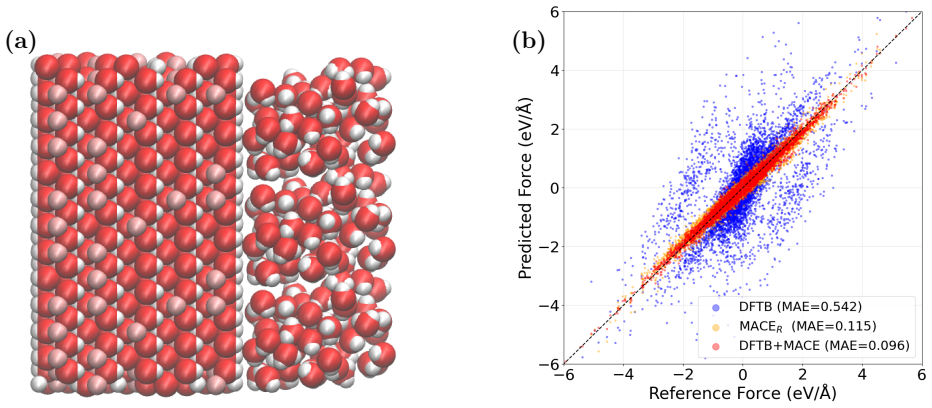
- Molecular dynamics of water clusters and phonon spectra of both fcc-MgO and bcc-MgO become feasible with better fidelity at moderate extra cost.
- Simulations can extend to length and time scales beyond direct DFT while an explicit electronic-structure description is retained.
- Charge-transfer processes in magnesium materials can be studied at scales inaccessible to pure first-principles methods.
Where Pith is reading between the lines
- The same residual-training strategy could be applied to other elemental or compound systems where a tight-binding baseline already exists.
- Expanding the original MACE training set to cover a wider range of magnesium configurations would be expected to reduce generalization errors in new simulations.
- The hybrid framework offers a practical route for studying defect migration or interface charge transfer in magnesium-based batteries or catalysts at intermediate scales.
Load-bearing premise
The MACE component trained on DFT minus DFTB differences will generalize accurately to the structural relaxations, molecular dynamics trajectories, and phonon calculations performed in the study.
What would settle it
Running the DFTB+MACE model on a set of magnesium structures outside the original training distribution and finding that its errors in energy or forces exceed those of standard DFTB plus pair potential would falsify the accuracy claim.
Figures
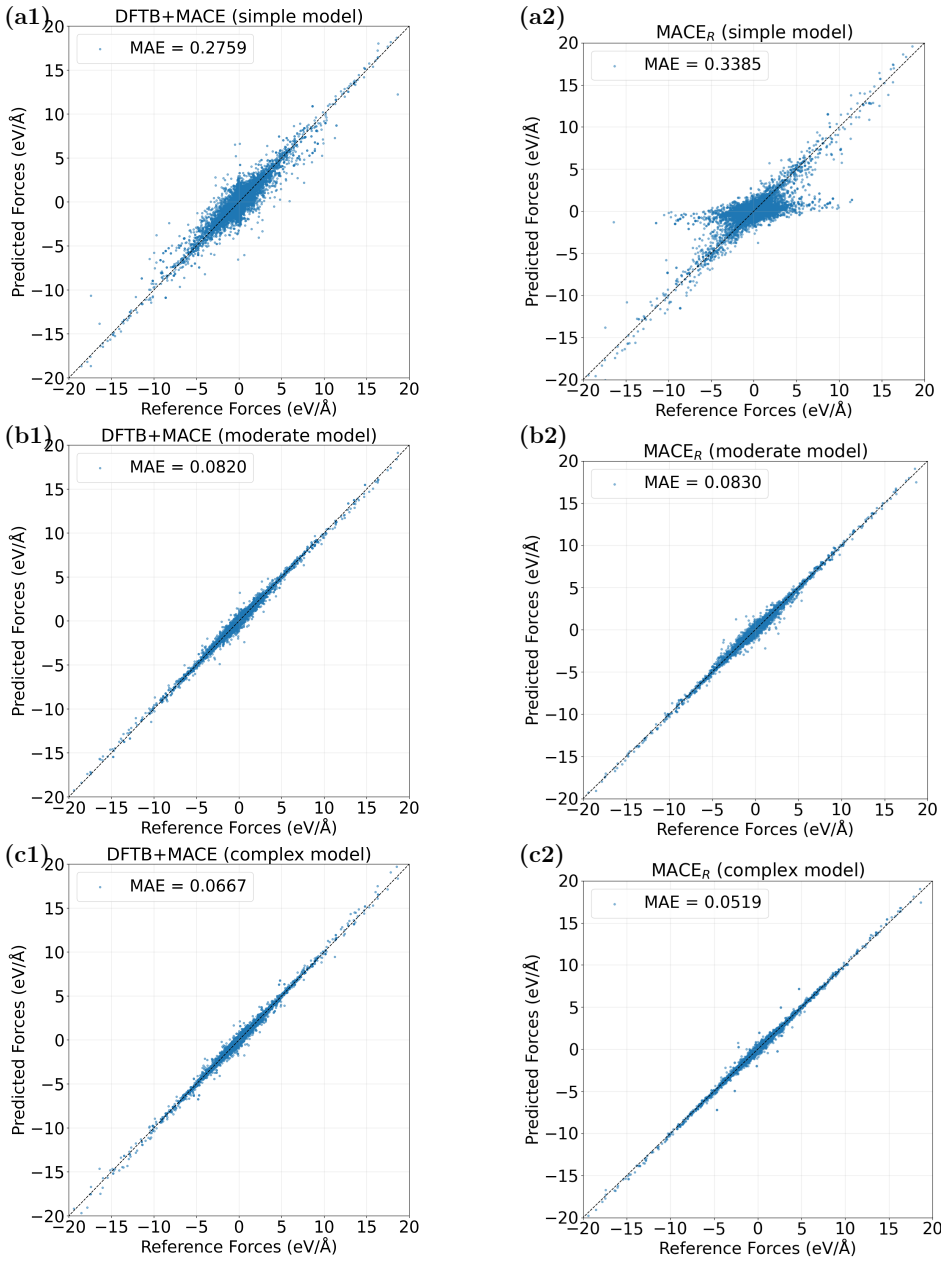
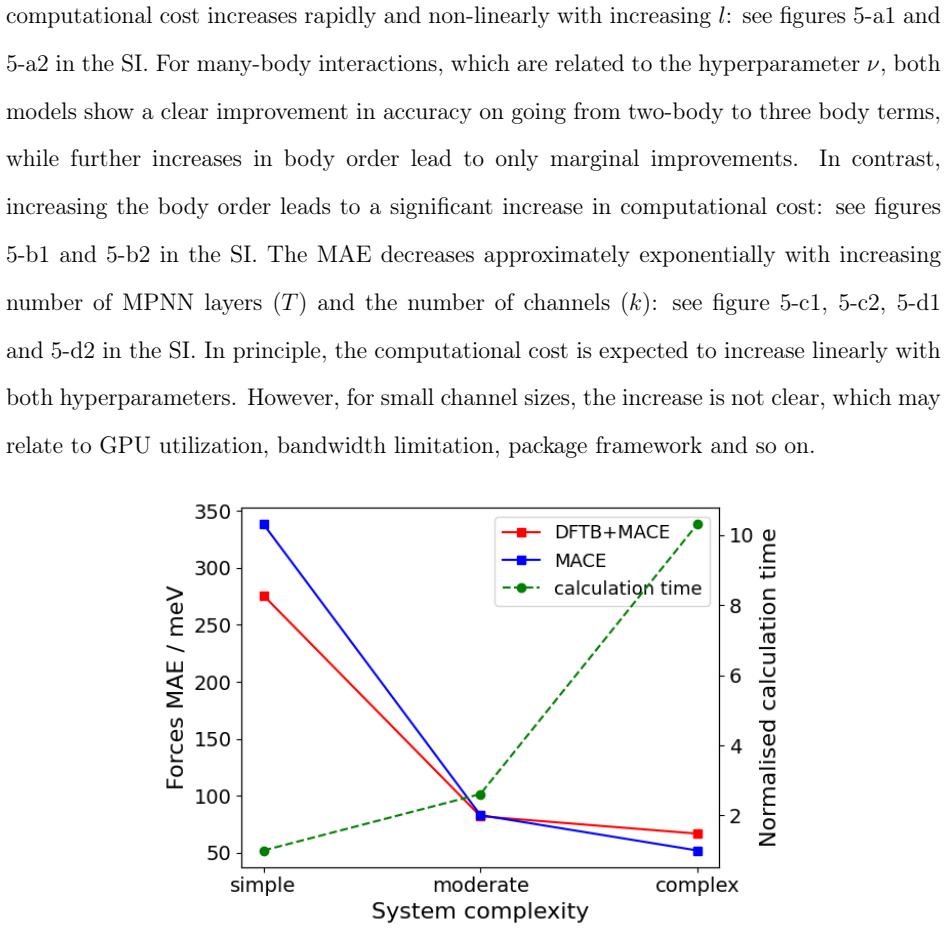
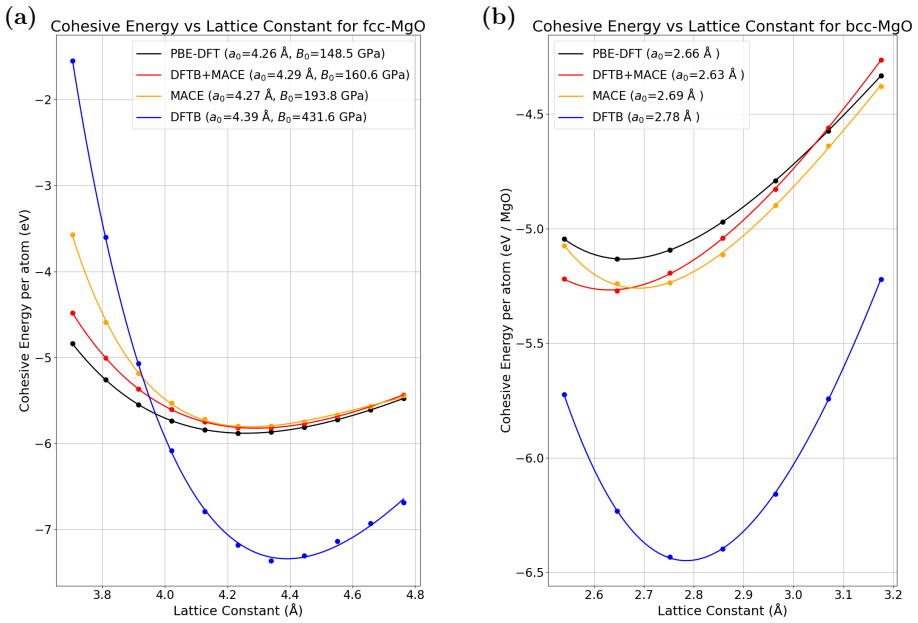
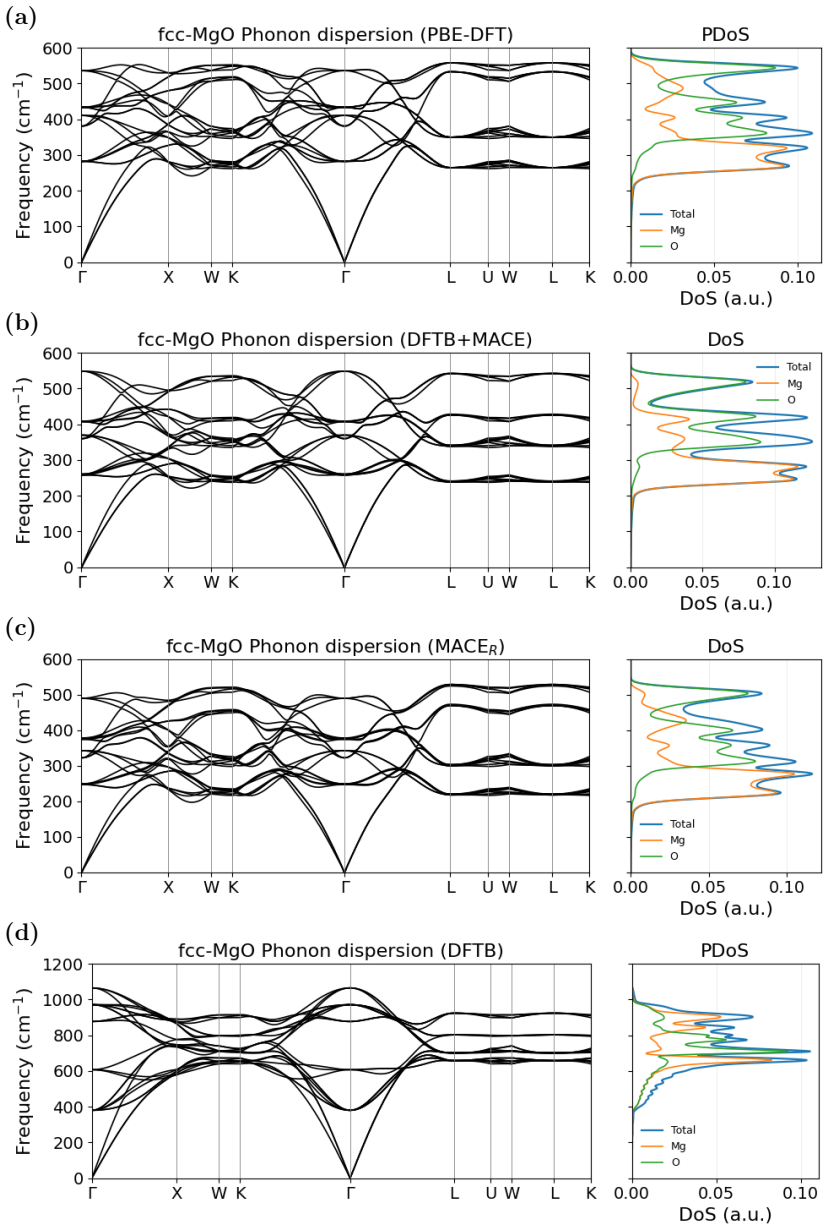
read the original abstract
We present a model for magnesium-based systems that combines density functional tight binding (DFTB) with MACE, a machine learning interatomic potential (DFTB+MACE). In this model, the conventional repulsive potential, pair potential, is replaced by a many-body MACE potential. The MACE component of the model is trained on the difference between density functional theory (DFT) energies and forces and the corresponding DFTB values, but neglecting the pair potential contribution. Using this model we performed structural relaxation of MgO-CO2 adsorption systems, molecular dynamics calculations of water clusters and phonon spectrum calculations of stable fcc-MgO and metastable bcc-MgO structures. We compare the performance of our model with a pure MACE model and with DFT. We demonstrate that the DFTB+MACE model achieves improved accuracy relative to DFTB with a pair potential, in many cases with only a moderate increase in computational cost. In addition, it can provide electronic structures that most of the machine learning potentials cannot. The training dataset, originally developed for MACE, may not fully represent all regions of the potential surface we may encounter during simulations. Expanding the dataset for a wider potential surface is expected to further enhance predictive accuracy of DFTB+MACE model. Overall, the resulting DFTB+MACE framework enables simulations at length and time scales beyond the reach of first-principles methods while retaining an explicit description of electronic structures, making it particularly attractive for studying charge-transfer in materials.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript introduces a hybrid DFTB+MACE model for magnesium-based systems in which the conventional repulsive pair potential of DFTB is replaced by a MACE many-body ML potential. The MACE component is trained on DFT minus DFTB energy/force differences (explicitly neglecting the pair-potential contribution). The model is applied to structural relaxations of MgO-CO2 adsorption systems, molecular-dynamics simulations of water clusters, and phonon calculations for stable fcc-MgO and metastable bcc-MgO; results are compared with pure MACE and DFT. The central claim is that DFTB+MACE improves accuracy relative to DFTB+pair potential, often at only moderate extra cost, while retaining an explicit electronic-structure description unavailable to most ML potentials.
Significance. If the claimed accuracy gains are shown to hold without uncontrolled extrapolation, the hybrid framework would usefully combine DFTB's electronic-structure access and linear-scaling potential with ML accuracy, enabling charge-transfer studies at length and time scales inaccessible to pure DFT. The approach is conceptually attractive for magnesium compounds, but its significance is currently limited by the absence of quantitative validation that the encountered configurations lie inside the training distribution.
major comments (2)
- [Abstract and Results (relaxations, MD, phonons)] Abstract (training description) and Results sections on relaxations/MD/phonons: the MACE component is trained on DFT-DFTB differences that neglect the pair potential, yet the manuscript itself states that the original MACE training set 'may not fully represent all regions of the potential surface we may encounter during simulations' of MgO-CO2, water clusters, and fcc/bcc-MgO phonons. No distribution-overlap test, nearest-neighbor analysis, or out-of-distribution error metric is reported to confirm that the local environments sampled in the reported calculations lie inside the training domain; this directly undermines the load-bearing claim of improved accuracy over DFTB+pair.
- [Results] Results (comparison statements): the claim that DFTB+MACE 'achieves improved accuracy relative to DFTB with a pair potential, in many cases with only a moderate increase in computational cost' is presented without tabulated RMSE values, error bars, or direct numerical comparisons against both DFTB+pair and pure MACE for the same observables (energies, forces, phonon frequencies). Without these data the central accuracy claim cannot be evaluated.
minor comments (1)
- [Abstract] Abstract: the final paragraph acknowledges the training-set limitation but offers only the generic remedy of 'expanding the dataset'; a concrete proposal (e.g., active-learning loop or targeted augmentation for water/MgO interfaces) would strengthen the discussion.
Simulated Author's Rebuttal
We thank the referee for the careful and constructive review. The comments highlight important points on validation and quantitative presentation that we address below. We agree that strengthening these aspects will improve the manuscript.
read point-by-point responses
-
Referee: Abstract (training description) and Results sections on relaxations/MD/phonons: the MACE component is trained on DFT-DFTB differences that neglect the pair potential, yet the manuscript itself states that the original MACE training set 'may not fully represent all regions of the potential surface we may encounter during simulations' of MgO-CO2, water clusters, and fcc/bcc-MgO phonons. No distribution-overlap test, nearest-neighbor analysis, or out-of-distribution error metric is reported to confirm that the local environments sampled in the reported calculations lie inside the training domain; this directly undermines the load-bearing claim of improved accuracy over DFTB+pair.
Authors: We acknowledge the manuscript already notes this limitation of the training set. No explicit distribution-overlap or nearest-neighbor analysis was performed in the original work. The reported comparisons to DFT and pure MACE provide supporting evidence of utility within the sampled regimes, but we agree that direct domain-overlap metrics would strengthen the presentation. We will add a nearest-neighbor analysis of the encountered configurations relative to the training set and a brief discussion of extrapolation risks in the revised manuscript. revision: yes
-
Referee: Results (comparison statements): the claim that DFTB+MACE 'achieves improved accuracy relative to DFTB with a pair potential, in many cases with only a moderate increase in computational cost' is presented without tabulated RMSE values, error bars, or direct numerical comparisons against both DFTB+pair and pure MACE for the same observables (energies, forces, phonon frequencies). Without these data the central accuracy claim cannot be evaluated.
Authors: The original manuscript presents comparisons primarily through figures rather than tabulated numerical summaries. We agree that explicit RMSE tables, error bars, and side-by-side numerical values for energies, forces, and phonon frequencies would make the accuracy claims more readily evaluable. We will add a summary table with these quantitative metrics (including direct comparisons to DFTB+pair and pure MACE) in the revised Results section. revision: yes
Circularity Check
No circularity: empirical training against external DFT data
full rationale
The paper presents DFTB+MACE as a hybrid model in which the MACE component is trained on the difference between external DFT energies/forces and DFTB values (explicitly neglecting the pair potential). All performance claims (improved accuracy over DFTB+pair, parity with pure MACE on relaxations/MD/phonons) are framed as outcomes of direct comparison to independent DFT reference calculations rather than any internal derivation or self-referential fit. No equations, self-citations, or uniqueness arguments are shown that would reduce the reported accuracy gains to quantities defined inside the paper; the procedure is a standard external-data training workflow and remains self-contained.
Axiom & Free-Parameter Ledger
Reference graph
Works this paper leans on
-
[1]
Physical review , volume=
Self-consistent equations including exchange and correlation effects , author=. Physical review , volume=. 1965 , publisher=
1965
-
[2]
Physical Review B , volume=
Construction of tight-binding-like potentials on the basis of density-functional theory: Application to carbon , author=. Physical Review B , volume=. 1995 , publisher=
1995
-
[3]
International journal of quantum chemistry , volume=
Calculations of molecules, clusters, and solids with a simplified LCAO-DFT-LDA scheme , author=. International journal of quantum chemistry , volume=. 1996 , publisher=
1996
-
[4]
Journal of Chemical Theory and Computation , volume=
Tight Binding Simulation of the MgO and Mg (OH) 2 Hydration and Carbonation Processes , author=. Journal of Chemical Theory and Computation , volume=. 2025 , publisher=
2025
-
[5]
Journal of chemical theory and computation , volume=
DFTB parameters for the periodic table: Part 1, electronic structure , author=. Journal of chemical theory and computation , volume=. 2013 , publisher=
2013
-
[6]
Journal of chemical theory and computation , volume=
DFTB parameters for the periodic table, part 2: Energies and energy gradients from hydrogen to calcium , author=. Journal of chemical theory and computation , volume=. 2015 , publisher=
2015
-
[7]
The Journal of Physical Chemistry Letters , volume=
Accurate many-body repulsive potentials for density-functional tight binding from deep tensor neural networks , author=. The Journal of Physical Chemistry Letters , volume=. 2020 , publisher=
2020
-
[8]
Chemical Science , volume=
Choosing the right molecular machine learning potential , author=. Chemical Science , volume=. 2021 , publisher=
2021
-
[9]
Physical review letters , volume=
Generalized neural-network representation of high-dimensional potential-energy surfaces , author=. Physical review letters , volume=. 2007 , publisher=
2007
-
[10]
Physical review letters , volume=
Gaussian approximation potentials: The accuracy of quantum mechanics, without the electrons , author=. Physical review letters , volume=. 2010 , publisher=
2010
-
[11]
Physical Review B , volume=
Atomic cluster expansion for accurate and transferable interatomic potentials , author=. Physical Review B , volume=. 2019 , publisher=
2019
-
[12]
Advances in neural information processing systems , volume=
MACE: Higher order equivariant message passing neural networks for fast and accurate force fields , author=. Advances in neural information processing systems , volume=
-
[13]
2025 , publisher=
The Design Space of E (3)-Equivariant Atom-Centered Interatomic Potentials , author=. 2025 , publisher=
2025
-
[14]
The Journal of Chemical Physics , volume=
Gaussian polarizable-ion tight binding , author=. The Journal of Chemical Physics , volume=. 2016 , publisher=
2016
-
[15]
Materials project trajectory (MPtrj) dataset , author=. Figshare. https://doi. org/10.6084/m9. figshare , volume=
-
[16]
arXiv preprint arXiv:2503.04070 , year=
A foundational potential energy surface dataset for materials , author=. arXiv preprint arXiv:2503.04070 , year=
-
[17]
Physical review letters , volume=
Generalized gradient approximation made simple , author=. Physical review letters , volume=. 1996 , publisher=
1996
-
[18]
Physical Review B , volume=
Separable dual-space Gaussian pseudopotentials , author=. Physical Review B , volume=. 1996 , publisher=
1996
-
[19]
Physical Review B , volume=
Relativistic separable dual-space Gaussian pseudopotentials from H to Rn , author=. Physical Review B , volume=. 1998 , publisher=
1998
-
[20]
Theoretical Chemistry Accounts , volume=
Pseudopotentials for H to Kr optimized for gradient-corrected exchange-correlation functionals , author=. Theoretical Chemistry Accounts , volume=. 2005 , publisher=
2005
-
[21]
Journal of physics: Condensed matter , volume=
QUANTUM ESPRESSO: a modular and open-source software project for quantumsimulations of materials , author=. Journal of physics: Condensed matter , volume=. 2009 , publisher=
2009
-
[22]
Physical review b , volume=
From ultrasoft pseudopotentials to the projector augmented-wave method , author=. Physical review b , volume=. 1999 , publisher=
1999
-
[23]
The Design Space of E(3)-Equivariant Atom-Centered Interatomic Potentials , author =. 2022 , number =. doi:10.48550/arXiv.2205.06643 , archiveprefix =. 2205.06643 , eprinttype =
-
[24]
Journal of computational chemistry , volume=
Accurate description of van der Waals complexes by density functional theory including empirical corrections , author=. Journal of computational chemistry , volume=. 2004 , publisher=
2004
-
[25]
The Journal of Chemical Physics , volume=
Enhancing the accuracy of density functional tight binding models through ChIMES many-body interaction potentials , author=. The Journal of Chemical Physics , volume=. 2023 , publisher=
2023
-
[26]
Journal of chemical theory and computation , volume=
Chimes: A force matched potential with explicit three-body interactions for molten carbon , author=. Journal of chemical theory and computation , volume=. 2017 , publisher=
2017
-
[27]
Physical Review B , volume=
Microscopic theory of force constants in the adiabatic approximation , author=. Physical Review B , volume=. 1970 , publisher=
1970
-
[28]
Journal of Chemical Theory and Computation , volume=
Curvature constrained splines for DFTB repulsive potential parametrization , author=. Journal of Chemical Theory and Computation , volume=. 2021 , publisher=
2021
-
[29]
The Journal of Physical Chemistry A , volume=
Initial Steps toward Automating the Fitting of DFTB E rep (r) , author=. The Journal of Physical Chemistry A , volume=. 2007 , publisher=
2007
-
[30]
Journal of chemical theory and computation , volume=
Tight-binding approximation-enhanced global optimization , author=. Journal of chemical theory and computation , volume=. 2018 , publisher=
2018
-
[31]
Journal of chemical theory and computation , volume=
Automated repulsive parametrization for the DFTB method , author=. Journal of chemical theory and computation , volume=. 2011 , publisher=
2011
-
[32]
The Journal of Physical Chemistry C , volume=
An SCC-DFTB repulsive potential for various ZnO polymorphs and the ZnO--water system , author=. The Journal of Physical Chemistry C , volume=. 2013 , publisher=
2013
-
[33]
The Journal of Physical Chemistry A , volume=
Recent developments in dftb+, a software package for efficient atomistic quantum mechanical simulations , author=. The Journal of Physical Chemistry A , volume=. 2025 , publisher=
2025
-
[34]
Journal of Chemical Theory and Computation , volume=
Semi-automated creation of density functional tight binding models through leveraging Chebyshev polynomial-based force fields , author=. Journal of Chemical Theory and Computation , volume=. 2021 , publisher=
2021
-
[35]
arXiv preprint arXiv:2207.09453 , year=
e3nn: Euclidean neural networks , author=. arXiv preprint arXiv:2207.09453 , year=
-
[36]
Journal of the American Chemical Society , volume=
The experimental determination of the surface tension of magnesium oxide , author=. Journal of the American Chemical Society , volume=. 1952 , publisher=
1952
-
[37]
Physical Chemistry Chemical Physics , volume=
First principles simulations of MgO (100) surface hydration at ambient conditions , author=. Physical Chemistry Chemical Physics , volume=. 2024 , publisher=
2024
-
[38]
Journal of Chemical Theory and Computation , volume=
Parametrization and benchmark of DFTB3 for organic molecules , author=. Journal of Chemical Theory and Computation , volume=. 2013 , publisher=
2013
-
[39]
Journal of chemical theory and computation , volume=
Parameterization of DFTB3/3OB for sulfur and phosphorus for chemical and biological applications , author=. Journal of chemical theory and computation , volume=. 2014 , publisher=
2014
-
[40]
The Journal of Physical Chemistry B , volume=
Parametrization of DFTB3/3OB for magnesium and zinc for chemical and biological applications , author=. The Journal of Physical Chemistry B , volume=. 2015 , publisher=
2015
-
[41]
Zeolites , volume=
Semi-relativistic, self-consistent charge Slater-Koster tables for density-functional based tight-binding (DFTB) for materials science simulations , author=. Zeolites , volume=
-
[42]
The Journal of Physical Chemistry C , volume=
Structural, electronic, and mechanical properties of single-walled chrysotile nanotube models , author=. The Journal of Physical Chemistry C , volume=. 2012 , publisher=
2012
-
[43]
Physical review E , volume=
Calculation of effective interaction potentials from radial distribution functions: A reverse Monte Carlo approach , author=. Physical review E , volume=. 1995 , publisher=
1995
-
[44]
Molecular Physics , volume=
In situ parameterisation of SCC-DFTB repulsive potentials by iterative Boltzmann inversion , author=. Molecular Physics , volume=. 2013 , publisher=
2013
-
[45]
The Journal of Physical Chemistry B , volume=
Molecular simulation of water and hydration effects in different environments: Challenges and developments for DFTB based models , author=. The Journal of Physical Chemistry B , volume=. 2014 , publisher=
2014
-
[46]
Journal of Chemical Theory and Computation , volume=
Accurate SCC-DFTB parametrization for bulk water , author=. Journal of Chemical Theory and Computation , volume=. 2020 , publisher=
2020
-
[47]
Nature Communications , volume=
Machine learning of charges and long-range interactions from energies and forces , author=. Nature Communications , volume=. 2025 , publisher=
2025
-
[48]
Nature Communications , volume=
A foundation machine learning potential with polarizable long-range interactions for materials modelling , author=. Nature Communications , volume=. 2025 , publisher=
2025
-
[49]
Physical Chemistry Chemical Physics , year=
Advancing Density Functional Tight-Binding method for Large Organic Molecules through Equivariant Neural Networks , author=. Physical Chemistry Chemical Physics , year=
-
[50]
Nature communications , volume=
SpookyNet: Learning force fields with electronic degrees of freedom and nonlocal effects , author=. Nature communications , volume=. 2021 , publisher=
2021
-
[51]
Nature Communications , volume=
Learning local equivariant representations for large-scale atomistic dynamics , author=. Nature Communications , volume=. 2023 , publisher=
2023
-
[52]
Computer Physics Communications , volume=
Efficient self-consistency for magnetic tight binding , author=. Computer Physics Communications , volume=. 2011 , publisher=
2011
-
[53]
The Journal of chemical physics , volume=
A foundation model for atomistic materials chemistry , author=. The Journal of chemical physics , volume=. 2025 , publisher=
2025
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.