Transferable Machine Learning of Electronic Hamiltonians with Superposition-of-Atomic-Potentials Features
Pith reviewed 2026-06-27 07:53 UTC · model grok-4.3
The pith
Superposition-of-atomic-potentials features let a graph neural network predict converged Kohn-Sham Fock matrices that transfer across molecules.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
Electronic features obtained from the superposition-of-atomic-potentials approximation already encode sufficient screening information that an orbital-based graph neural network can learn a direct mapping to converged Kohn-Sham Fock matrices, producing accurate electronic-structure quantities that remain reliable when applied to new molecular systems and when downfolded to larger basis sets.
What carries the argument
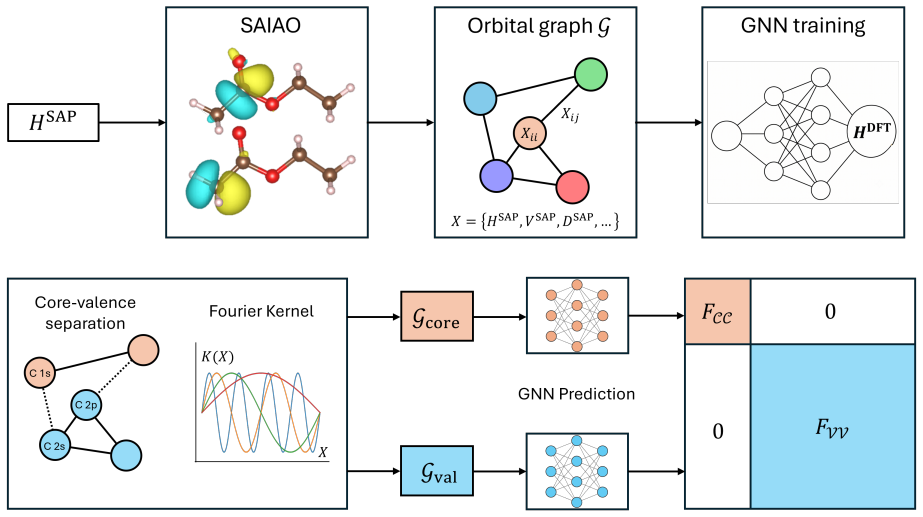
The superposition-of-atomic-potentials (SAP) approximation, which defines a symmetry-adapted intrinsic atomic orbital learning basis and supplies the physics-informed inputs to the orbital-based graph neural network that predicts the Fock matrix.
If this is right
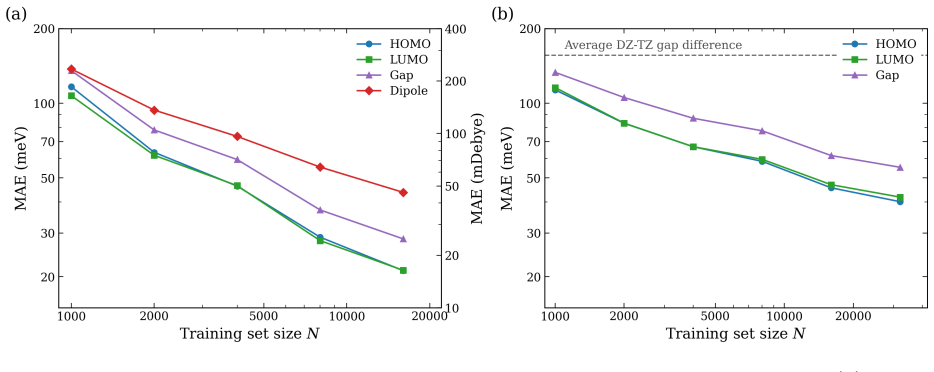
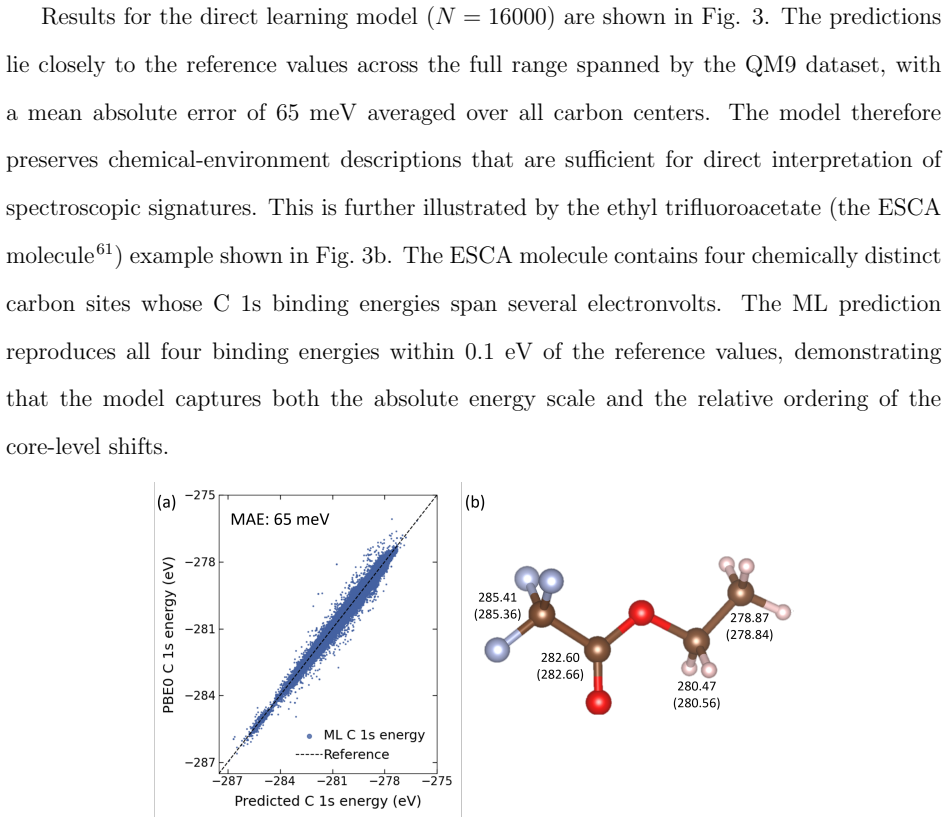
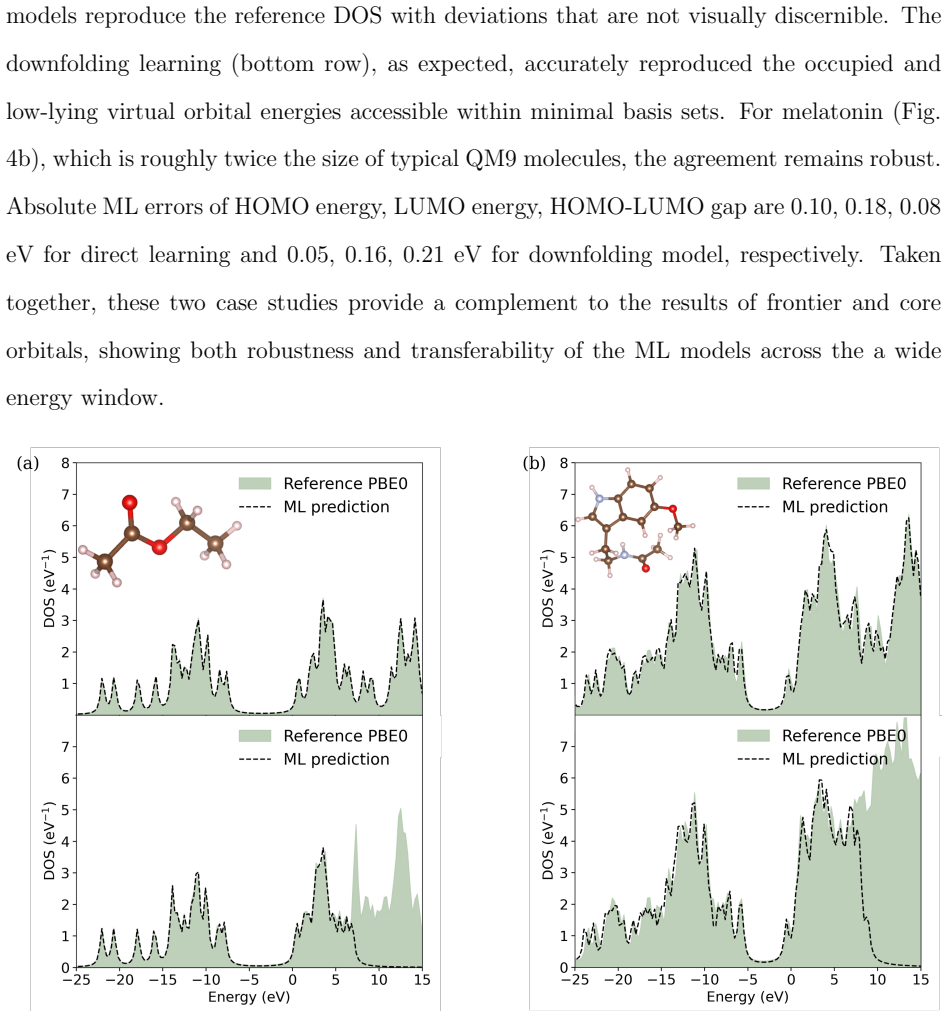
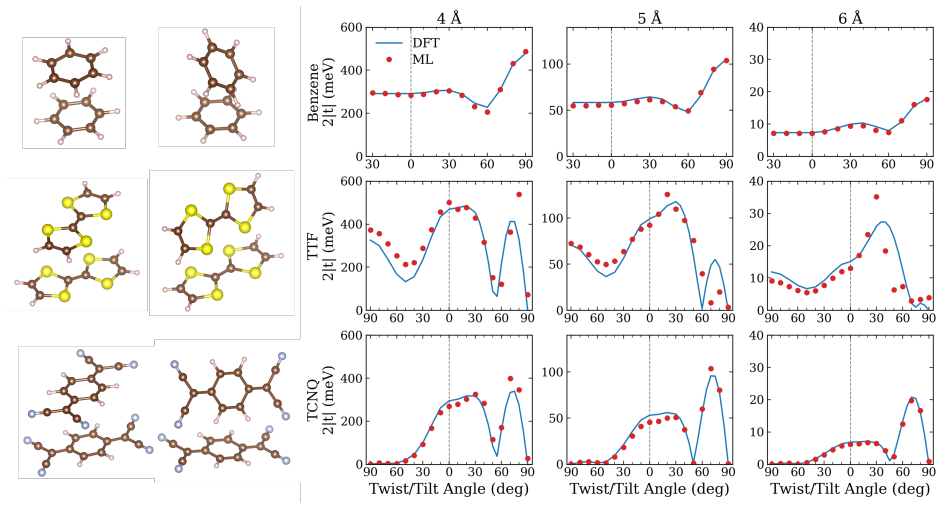
- Frontier and core orbital energies, dipole moments, and the full density of states are reproduced on the QM9 dataset.
- Intermolecular transfer integrals are obtained accurately for benzene, TCNQ, and TTF dimers.
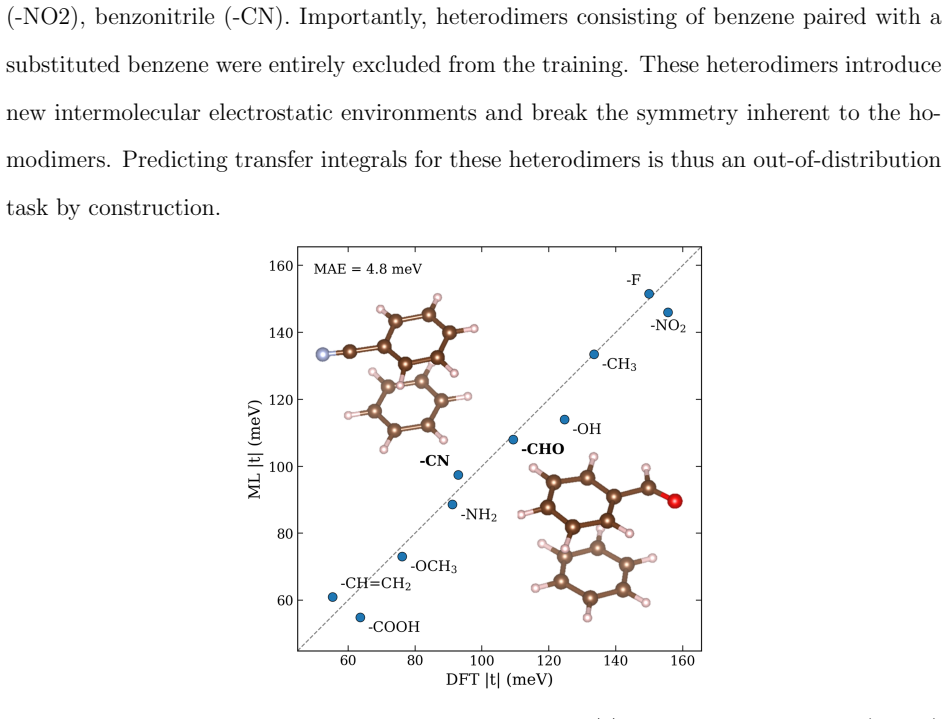
- Predictions transfer to unseen substituted-benzene heterodimers at a mean absolute error of 4.8 meV.
- A downfolding procedure extends the predictions from minimal-basis features to larger basis sets.
Where Pith is reading between the lines
- High-throughput screening of charge-transport materials becomes feasible without repeated self-consistent-field calculations for each new candidate.
- The same SAP-derived features could be tested for predicting response properties such as polarizabilities once the Fock matrix is available.
- Extension to periodic systems would require only redefinition of the atomic-orbital basis to respect translational symmetry.
Load-bearing premise
The superposition-of-atomic-potentials approximation already supplies features that capture the essential electron-electron screening required for the network to map reliably to converged Fock matrices.
What would settle it
Compute the model's predicted Fock matrix and resulting transfer integrals for a dimer whose SAP initial guess deviates strongly from the converged solution; large errors would indicate that the learned mapping does not hold when screening is not adequately captured.
Figures
read the original abstract
Machine learning (ML) of electronic Hamiltonians offers a unified route to electronic wave functions and physical observables. We introduce a Hamiltonian learning framework built on electronic features derived from the superposition-of-atomic-potentials (SAP) approximation, an efficient self-consistent-field initial guess that captures essential electron-electron screening. SAP quantities define a symmetry-adapted intrinsic atomic orbital learning basis and provide physics-informed inputs to an orbital-based graph neural network that predicts converged Kohn-Sham Fock matrices. To extend the approach to larger basis sets, we further develop a downfolding scheme that predicts large-basis electronic structure from minimal-basis features. On the QM9 dataset, the model accurately reproduces frontier and core orbital energies, dipole moments, and the full density of states. For organic charge-transport materials, it yields accurate intermolecular transfer integrals for benzene, tetracyanoquinodimethane (TCNQ), and tetrathiafulvalene (TTF) dimers, and transfers to unseen substituted-benzene heterodimers with a mean absolute error of 4.8 meV. These results establish SAP-based ML of electronic Hamiltonians as a transferable and scalable tool for high-throughput electronic-structure prediction.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript introduces a machine learning framework for predicting electronic Hamiltonians, using features from the superposition-of-atomic-potentials (SAP) approximation to define a symmetry-adapted intrinsic atomic orbital (IAO) basis and to supply physics-informed inputs to an orbital-based graph neural network (GNN) that outputs converged Kohn-Sham Fock matrices. A downfolding scheme is developed to extend predictions from minimal to larger basis sets. Results are reported on the QM9 dataset for frontier/core orbital energies, dipole moments, and density of states, as well as on organic charge-transport dimers (benzene, TCNQ, TTF) for intermolecular transfer integrals, with claimed transfer to unseen substituted-benzene heterodimers at 4.8 meV MAE.
Significance. If the central results hold after addressing validation gaps, the work would offer a scalable route to transferable Hamiltonian prediction that combines efficient mean-field features with GNNs, potentially reducing the cost of high-throughput electronic-structure calculations for molecular materials while preserving physical observables such as transfer integrals.
major comments (2)
- [Abstract and Results] Abstract and Results sections: numerical accuracies (e.g., 4.8 meV MAE on heterodimers) are stated without any description of training/validation splits, cross-validation protocol, error bars, or baseline comparisons (geometry-only GNN or standard SAP guess). This information is required to judge whether the reported transferability is supported by the data and is load-bearing for the claim that the model generalizes to unseen substituted-benzene heterodimers.
- [Methods (SAP feature construction and GNN input)] Methods (SAP feature construction and GNN input): the central assumption that SAP quantities already encode the essential molecule-specific electron-electron screening (so that the GNN learns a reliable mapping to converged Fock matrices) is not isolated by ablation. Without a direct comparison of SAP+GNN versus geometry-only inputs on the heterodimer transfer task, it remains possible that the network is fitting data-specific corrections rather than leveraging transferable physics; this directly affects the claimed transferability and downfolding scheme.
minor comments (2)
- [Methods] Clarify in the text how the symmetry-adapted IAO basis is exactly constructed from SAP quantities and whether any additional orthogonalization steps are performed.
- [Results] Add learning curves or training-set-size dependence to demonstrate that the reported MAEs are not artifacts of a particular data partition.
Simulated Author's Rebuttal
We thank the referee for the careful reading and constructive comments. We address each major point below and have revised the manuscript to supply the requested details and comparisons.
read point-by-point responses
-
Referee: [Abstract and Results] Abstract and Results sections: numerical accuracies (e.g., 4.8 meV MAE on heterodimers) are stated without any description of training/validation splits, cross-validation protocol, error bars, or baseline comparisons (geometry-only GNN or standard SAP guess). This information is required to judge whether the reported transferability is supported by the data and is load-bearing for the claim that the model generalizes to unseen substituted-benzene heterodimers.
Authors: We agree that the original manuscript did not provide sufficient detail on data splits, validation protocol, error bars, or baselines. In the revised version we have added an explicit subsection in Methods describing the train/validation/test partitioning for QM9 and the dimer sets, the cross-validation procedure, standard deviations over five independent runs, and direct numerical comparisons against both a geometry-only GNN and the plain SAP guess. These additions show that the 4.8 meV MAE on the held-out substituted-benzene heterodimers is obtained under a strict unseen-molecule protocol and is meaningfully lower than the baselines. revision: yes
-
Referee: [Methods (SAP feature construction and GNN input)] Methods (SAP feature construction and GNN input): the central assumption that SAP quantities already encode the essential molecule-specific electron-electron screening (so that the GNN learns a reliable mapping to converged Fock matrices) is not isolated by ablation. Without a direct comparison of SAP+GNN versus geometry-only inputs on the heterodimer transfer task, it remains possible that the network is fitting data-specific corrections rather than leveraging transferable physics; this directly affects the claimed transferability and downfolding scheme.
Authors: We accept that an explicit ablation on the heterodimer transfer task is needed to isolate the contribution of the SAP features. We have performed this comparison and report the results in the revised Methods and Results sections: the SAP+GNN model achieves 4.8 meV MAE while the geometry-only variant yields ~25 meV MAE on the same unseen heterodimer set. This quantitative gap supports that the model is leveraging the physics-informed screening encoded in the SAP quantities rather than merely memorizing data-specific corrections, and we have clarified the implications for the downfolding procedure. revision: yes
Circularity Check
No circularity: trained mapping from independent SAP features to Fock matrices
full rationale
The paper presents a standard supervised ML pipeline in which SAP-derived quantities serve as fixed, physics-informed input features to a GNN that is trained to output converged KS Fock matrices. Reported performance is measured on held-out molecules (QM9) and transfer tasks (substituted-benzene heterodimers) against external reference calculations; no equation, definition, or self-citation is shown to make the target output identical to the input by construction. The derivation chain therefore remains non-circular and externally benchmarked.
Axiom & Free-Parameter Ledger
Reference graph
Works this paper leans on
-
[1]
T.; Davies, D
(1) Butler, K. T.; Davies, D. W.; Cartwright, H.; Isayev, O.; Walsh, A. Machine Learning for Molecular and Materials Science.Nature2018,559, 547–555. (2) von Lilienfeld, O. A.; M¨ uller, K.-R.; Tkatchenko, A. Exploring Chemical Compound Space with Quantum-Based Machine Learning.Nat. Rev. Chem.2020,4, 347–358. (3) Keith, J. A.; Vassilev-Galindo, V.; Cheng,...
2020
-
[2]
J.; Rabani, E
(6) Lin, K.; Coley-O’Rourke, M. J.; Rabani, E. Deep-Learning Atomistic Semi-Empirical Pseudopotential Model for Nanomaterials.npj Comput. Mater.2025,11,
2025
-
[3]
S.; Grzenda, D.; Zhu, R.; Hudson, N.; Foster, I.; Cheng, B.; Gagliardi, L
(7) King, D. S.; Grzenda, D.; Zhu, R.; Hudson, N.; Foster, I.; Cheng, B.; Gagliardi, L. Cartesian Equivariant Representations for Learning and Understanding Molecular Or- bitals.Proc. Natl. Acad. Sci.2025,122, e2510235122. (8) Behler, J.; Parrinello, M. Generalized Neural-Network Representation of High- Dimensional Potential-Energy Surfaces.Phys. Rev. Let...
2025
-
[4]
(12) Batatia, I.; Benner, P.; Chiang, Y.; Elena, A. M.; Kov´ acs, D. P.; Riebesell, J.; Ad- vincula, X. R.; Asta, M.; Avaylon, M.; Baldwin, W. J.; Berger, F.; Bernstein, N.; Bhowmik, A.; Bigi, F.; Blau, S. M.; C˘ arare, V.; Ceriotti, M.; Chong, S.; Darby, J. P.; De, S.; Della Pia, F.; Deringer, V. L.; Elijoˇ sius, R.; El-Machachi, Z.; Fako, E.; Fal- cioni...
arXiv 2025
-
[5]
(19) Westermayr, J.; J. Maurer, R. Physically Inspired Deep Learning of Molecular Excita- tions and Photoemission Spectra.Chem. Sci.2021,12, 10755–10764. (20) Cignoni, E.; Suman, D.; Nigam, J.; Cupellini, L.; Mennucci, B.; Ceriotti, M. Electronic Excited States from Physically Constrained Machine Learning.ACS Cent. Sci.2024, 10, 637–648. (21) Hou, B.; Xu,...
arXiv 2021
-
[6]
M.; Corminboeuf, C.; Ceriotti, M
(25) Grisafi, A.; Fabrizio, A.; Meyer, B.; Wilkins, D. M.; Corminboeuf, C.; Ceriotti, M. Transferable Machine-Learning Model of the Electron Density.ACS Cent. Sci.2019, 5, 57–64. (26) Shao, X.; Paetow, L.; Tuckerman, M. E.; Pavanello, M. Machine Learning Electronic Structure Methods Based on the One-Electron Reduced Density Matrix.Nat. Commun. 2023,14,
2019
-
[7]
(27) Hou, B.; Wu, J.; Qiu, D. Y. Unsupervised Representation Learning of Kohn–Sham States and Consequences for Downstream Predictions of Many-Body Effects.Nat. Com- mun.2024,15,
2024
-
[8]
(28) Rath, Y.; Booth, G. H. Interpolating Numerically Exact Many-Body Wave Functions for Accelerated Molecular Dynamics.Nat. Commun.2025,16,
2025
-
[9]
Deep-Learning Density Functional Theory Hamiltonian for Efficient Ab Initio Electronic-Structure Calculation.Nat
(29) Li, H.; Wang, Z.; Zou, N.; Ye, M.; Xu, R.; Gong, X.; Duan, W.; Xu, Y. Deep-Learning Density Functional Theory Hamiltonian for Efficient Ab Initio Electronic-Structure Calculation.Nat. Comput. Sci.2022,2, 367–377. (30) Nigam, J.; Willatt, M. J.; Ceriotti, M. Equivariant Representations for Molecular Hamiltonians and N-center Atomic-Scale Properties.J....
2022
-
[10]
K.; Zhang, P.; Zhang, L.; E, W
(33) Gu, Q.; Zhouyin, Z.; Pandey, S. K.; Zhang, P.; Zhang, L.; E, W. Deep Learning Tight- Binding Approach for Large-Scale Electronic Simulations at Finite Temperatures with Ab Initio Accuracy.Nat. Commun.2024,15,
2024
-
[11]
Machine Learning Many-Body Green’s Functions for Molecular Excitation Spectra.J
(34) Venturella, C.; Hillenbrand, C.; Li, J.; Zhu, T. Machine Learning Many-Body Green’s Functions for Molecular Excitation Spectra.J. Chem. Theory Comput.2024,20, 143–
2024
-
[12]
(35) Yu, H.; Liu, M.; Luo, Y.; Strasser, A.; Qian, X.; Qian, X.; Ji, S. QH9: A Quan- tum Hamiltonian Prediction Benchmark for QM9 Molecules.arXiv preprint2024, arXiv:2306.09549. (36) Suman, D.; Nigam, J.; Saade, S.; Pegolo, P.; T¨ urk, H.; Zhang, X.; Chan, G. K.-L.; Ceriotti, M. Exploring the Design Space of Machine Learning Models for Quantum Chemistry w...
arXiv 2025
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.