Retrieval-Augmented Multimodal Learning for Enzyme-Substrate Interaction Prediction Under Low-Homology Shift
Pith reviewed 2026-06-26 09:17 UTC · model grok-4.3
The pith
Retrieval of neighboring enzymes improves substrate interaction predictions when test sequences share little identity with training data.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
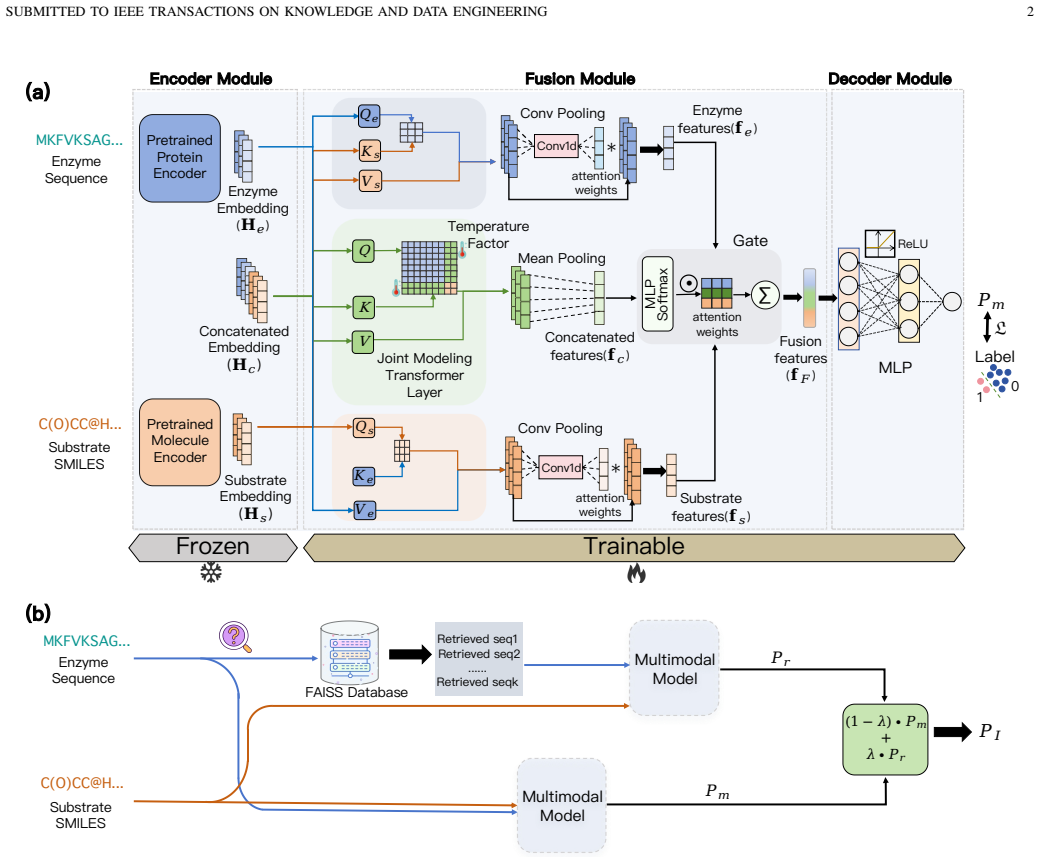
RAMMESI learns explicit pairwise enzyme-substrate representations through directional cross-modal interaction modeling and adaptive fusion; at inference it retrieves neighboring enzymes, recombines each with the query substrate, aggregates the pairwise predictions as contextual evidence, and applies an imbalance-aware weighted-BCE objective, yielding stronger performance on ESI benchmarks under sequence-identity-aware splits especially in low-identity regimes while improving multiple ESI backbones in a plug-and-play manner.
What carries the argument
The retrieval module that retrieves neighboring enzymes at inference time, recombines them with the query substrate, and aggregates the resulting pairwise predictions as contextual evidence.
If this is right
- Retrieval supplies a general mechanism that can be added to existing ESI backbones to increase robustness under homology shift.
- Explicit pairwise representations combined with adaptive fusion capture directional enzyme-substrate interactions more effectively than unimodal baselines.
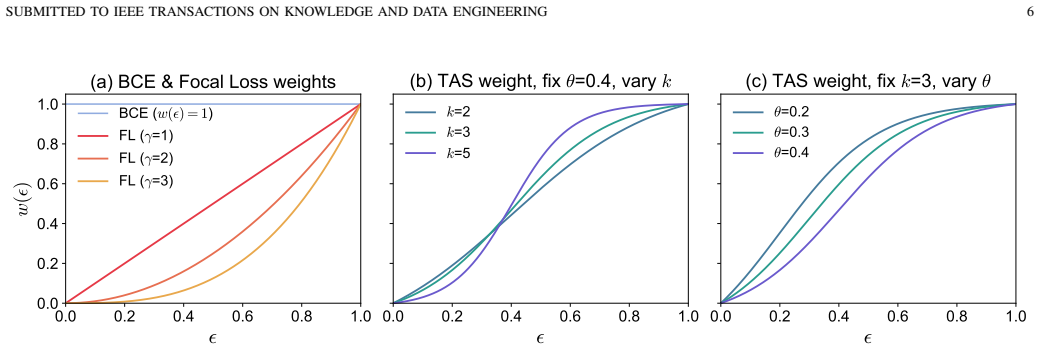
- The imbalance-aware weighted-BCE objective mitigates the effect of sparse positive supervision on learning.
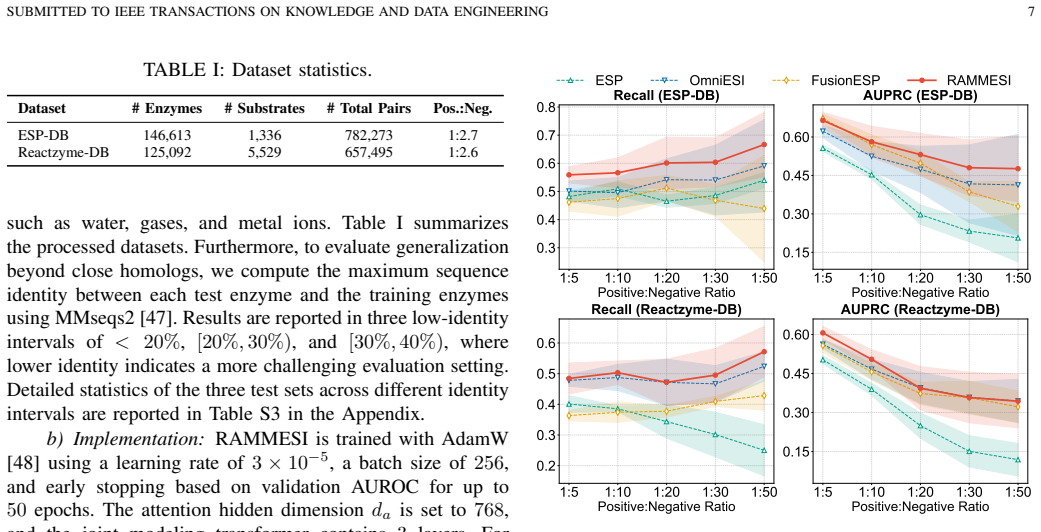
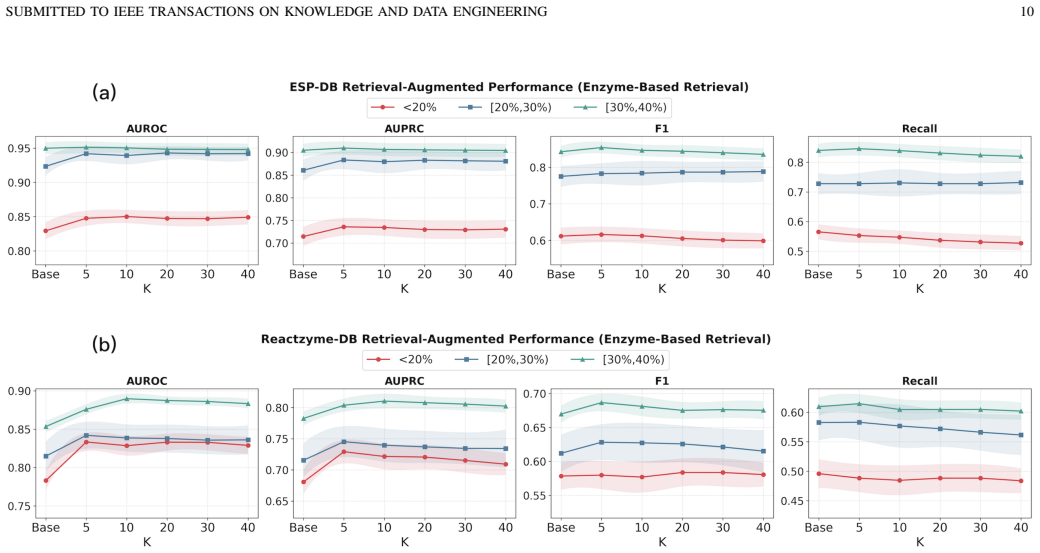
- Performance advantages grow as sequence identity between train and test enzymes decreases, enabling more reliable screening in large biochemical spaces.
Where Pith is reading between the lines
- The same retrieval-at-inference pattern could be tested on other protein-ligand tasks that suffer from homology shift, such as predicting binding for novel protein families.
- If the retrieved neighbors are drawn from public enzyme databases, the approach may reduce the labeled data needed to screen new biocatalysts.
- Extending the retrieval step to also pull in substrate analogs might further improve generalization when both enzyme and substrate novelty are high.
Load-bearing premise
Retrieving neighboring enzymes at inference and aggregating their predictions supplies useful contextual evidence that improves robustness under low-homology shift rather than merely averaging correlated errors.
What would settle it
An experiment showing that replacing retrieved neighbors with random enzymes produces the same accuracy gain on low-identity test sets, or that removing the retrieval step leaves low-identity performance unchanged.
Figures

read the original abstract
Enzyme substrate interaction (ESI) prediction is a fundamental computational task for biocatalyst discovery and reaction screening in large biochemical spaces. In practical settings, ESI prediction is challenged by sparse positive supervision and low-homology distribution shift, where test enzymes share limited sequence identity with those observed during training. To address these challenges, we propose RAMMESI, a retrieval-augmented multimodal framework for robust ESI prediction. RAMMESI learns explicit pairwise enzyme-substrate representations through directional cross-modal interaction modeling and adaptive fusion. To enhance robustness, RAMMESI retrieves neighboring enzymes at inference time, recombines them with the query substrate, and aggregates the resulting pairwise predictions as contextual evidence. To improve learning under sparse positive supervision, we further adopt an imbalance-aware weighted-BCE objective. Experiments on two ESI benchmarks under sequence-identity-aware splits demonstrate that RAMMESI achieves consistently strong performance, with particular advantages in more challenging low-identity regimes. In addition, the retrieval module improves multiple ESI backbones in a plug-and-play manner, suggesting that retrieval provides a general mechanism for improving robustness under homology shift.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The paper proposes RAMMESI, a retrieval-augmented multimodal framework for enzyme-substrate interaction (ESI) prediction. It learns explicit pairwise enzyme-substrate representations via directional cross-modal interaction modeling and adaptive fusion, employs an imbalance-aware weighted-BCE loss for sparse supervision, and at inference retrieves neighboring enzymes, recombines them with the query substrate, and aggregates the resulting pairwise predictions to improve robustness under low-homology distribution shift. Experiments on two ESI benchmarks using sequence-identity-aware splits report consistently strong performance with particular gains in low-identity regimes; the retrieval module is shown to improve multiple ESI backbones in a plug-and-play manner.
Significance. If the central claims hold after addressing mechanism-isolation concerns, the work would provide a practical, general mechanism for robustness under homology shift in ESI prediction, which is relevant for biocatalyst discovery. The use of sequence-identity-aware splits and the plug-and-play demonstration on multiple backbones are concrete strengths that would support broader applicability if the gains are shown to arise from contextual evidence rather than variance reduction.
major comments (2)
- [§3.3 and §5] The central claim that retrieval supplies useful contextual evidence improving robustness specifically under low-homology shift (abstract; §3.3 retrieval module; §5 experiments) is load-bearing yet lacks controls to distinguish this from averaging of correlated errors. The manuscript should specify the retrieval criterion (sequence identity, embedding similarity, or other), the source pool for neighbors, and include ablations such as random retrieval, retrieval from functionally unrelated enzymes, or comparison against simple ensemble averaging; without these, reported advantages on sequence-identity-aware splits could be explained by statistical effects rather than shift robustness.
- [§5] Table 2 (or equivalent results table) and the plug-and-play experiments: the reported gains on multiple backbones must be accompanied by statistical tests (e.g., paired t-tests or Wilcoxon across runs) and ablation of the aggregation function; current presentation leaves open whether improvements are consistent or driven by particular backbones or identity bins.
minor comments (2)
- [Abstract] Abstract: while the high-level claims are clear, a single sentence summarizing the key quantitative improvements (e.g., average AUC or top-k recall deltas versus baselines in the <30% identity regime) would improve readability without lengthening the abstract.
- [§3.2] Notation in §3.2: the directional cross-modal interaction and adaptive fusion modules would benefit from an explicit equation for the final fused representation before the prediction head, to make the multimodal component self-contained.
Simulated Author's Rebuttal
We thank the referee for the constructive feedback. We address each major comment below and will incorporate the requested controls, specifications, and statistical analyses in the revised manuscript to strengthen the evidence for the retrieval module.
read point-by-point responses
-
Referee: [§3.3 and §5] The central claim that retrieval supplies useful contextual evidence improving robustness specifically under low-homology shift (abstract; §3.3 retrieval module; §5 experiments) is load-bearing yet lacks controls to distinguish this from averaging of correlated errors. The manuscript should specify the retrieval criterion (sequence identity, embedding similarity, or other), the source pool for neighbors, and include ablations such as random retrieval, retrieval from functionally unrelated enzymes, or comparison against simple ensemble averaging; without these, reported advantages on sequence-identity-aware splits could be explained by statistical effects rather than shift robustness.
Authors: We agree that isolating the contribution of contextual evidence from statistical averaging is important. The retrieval criterion in RAMMESI is sequence identity (to match the sequence-identity-aware splits), with neighbors drawn from the training-set enzyme pool. In the revision we will explicitly state this in §3.3 and add the requested ablations in §5: (i) random retrieval, (ii) retrieval restricted to functionally unrelated enzymes (EC-number mismatch), and (iii) comparison against simple ensemble averaging of multiple independent predictions. These controls will demonstrate that performance gains arise from relevant neighbors rather than variance reduction alone. revision: yes
-
Referee: [§5] Table 2 (or equivalent results table) and the plug-and-play experiments: the reported gains on multiple backbones must be accompanied by statistical tests (e.g., paired t-tests or Wilcoxon across runs) and ablation of the aggregation function; current presentation leaves open whether improvements are consistent or driven by particular backbones or identity bins.
Authors: We acknowledge the need for statistical rigor and further dissection of the aggregation step. In the revised §5 we will report paired t-tests (or Wilcoxon signed-rank tests) across independent runs for all plug-and-play results on multiple backbones. We will also add an ablation of the aggregation function (mean vs. weighted or attention-based aggregation) and present results broken down by sequence-identity bins to confirm that gains are consistent rather than driven by specific backbones or bins. revision: yes
Circularity Check
No significant circularity; empirical framework validated on benchmarks
full rationale
The paper introduces RAMMESI as a retrieval-augmented multimodal model for ESI prediction and reports its performance on two benchmarks under sequence-identity-aware splits. No derivation chain, equations, or first-principles results are presented that reduce to fitted inputs or self-citations. The retrieval mechanism is described as a plug-and-play addition whose benefit is shown experimentally rather than derived by construction from the inputs. The central claims rest on observed performance improvements rather than any self-referential loop, making the work self-contained against external benchmarks.
Axiom & Free-Parameter Ledger
Reference graph
Works this paper leans on
-
[1]
Industrial applications of enzymes: Recent advances, techniques, and outlooks,
J. Chapman, A. E. Ismail, and C. Z. Dinu, “Industrial applications of enzymes: Recent advances, techniques, and outlooks,”Catalysts, vol. 8, no. 6, p. 238, 2018
2018
-
[2]
From nature to industry: Harnessing enzymes for biocatalysis,
R. Buller, S. Lutz, R. Kazlauskas, R. Snajdrova, J. Moore, and U. Born- scheuer, “From nature to industry: Harnessing enzymes for biocatalysis,” Science, vol. 382, no. 6673, p. eadh8615, 2023
2023
-
[3]
Enzyme function prediction using contrastive learning,
T. Yu, H. Cui, J. C. Li, Y . Luo, G. Jiang, and H. Zhao, “Enzyme function prediction using contrastive learning,”Science, vol. 379, no. 6639, pp. 1358–1363, 2023
2023
-
[4]
Accelerating enzyme discovery and engineering with high-throughput screening,
E. U. Bozkurt, E. C. Ørsted, D. C. V olke, and P. I. Nikel, “Accelerating enzyme discovery and engineering with high-throughput screening,” Natural Product Reports, 2026
2026
-
[5]
Uniprot: the universal protein knowledgebase in 2025,
“Uniprot: the universal protein knowledgebase in 2025,”Nucleic Acids Research, vol. 53, no. D1, pp. D609–D617, 2025
2025
-
[6]
Combining structure and sequence information allows automated prediction of substrate speci- ficities within enzyme families,
M. R ¨ottig, C. Rausch, and O. Kohlbacher, “Combining structure and sequence information allows automated prediction of substrate speci- ficities within enzyme families,”PLoS Computational Biology, vol. 6, no. 1, p. e1000636, 2010
2010
-
[7]
Predicting novel substrates for enzymes with minimal ex- perimental effort with active learning,
D. A. Pertusi, M. E. Moura, J. G. Jeffryes, S. Prabhu, B. W. Biggs, and K. E. Tyo, “Predicting novel substrates for enzymes with minimal ex- perimental effort with active learning,”Metabolic Engineering, vol. 44, pp. 171–181, 2017
2017
-
[8]
Functional and informatics analysis enables glycosyltransferase activity prediction,
M. Yang, C. Fehl, K. V . Lees, E.-K. Lim, W. A. Offen, G. J. Davies, D. J. Bowles, M. G. Davidson, S. J. Roberts, and B. G. Davis, “Functional and informatics analysis enables glycosyltransferase activity prediction,” Nature Chemical Biology, vol. 14, no. 12, pp. 1109–1117, 2018
2018
-
[9]
Machine learning-based prediction of enzyme substrate scope: application to bacterial nitrilases,
Z. Mou, J. Eakes, C. J. Cooper, C. M. Foster, R. F. Standaert, M. Podar, M. J. Doktycz, and J. M. Parks, “Machine learning-based prediction of enzyme substrate scope: application to bacterial nitrilases,”Proteins: Structure, Function, and Bioinformatics, vol. 89, no. 3, pp. 336–347, 2021
2021
-
[10]
Enzymclass: Substrate specificity prediction tool of plant acyl-acp thioesterases based on ensemble learning,
D. Banerjee, M. A. Jindra, A. J. Linot, B. F. Pfleger, and C. D. Maranas, “Enzymclass: Substrate specificity prediction tool of plant acyl-acp thioesterases based on ensemble learning,”Current Research in Biotechnology, vol. 4, pp. 1–9, 2022
2022
-
[11]
A general model to predict small molecule substrates of enzymes based on machine and deep learning,
A. Kroll, S. Ranjan, M. K. Engqvist, and M. J. Lercher, “A general model to predict small molecule substrates of enzymes based on machine and deep learning,”Nature Communications, vol. 14, no. 1, p. 2787, 2023
2023
-
[12]
GIaNt: Protein-ligand binding affinity prediction via geometry-aware interactive graph neural network,
S. Li, J. Zhou, T. Xu, L. Huang, F. Wang, H. Xiong, W. Huang, D. Dou, and H. Xiong, “GIaNt: Protein-ligand binding affinity prediction via geometry-aware interactive graph neural network,”IEEE Transactions on Knowledge and Data Engineering, vol. 36, no. 5, pp. 1991–2008, 2023
1991
-
[13]
KG-MTL: knowledge graph enhanced multi-task learning for molecular interaction,
T. Ma, X. Lin, B. Song, P. S. Yu, and X. Zeng, “KG-MTL: knowledge graph enhanced multi-task learning for molecular interaction,”IEEE Transactions on Knowledge and Data Engineering, vol. 35, no. 7, pp. 7068–7081, 2022
2022
-
[14]
Learning to denoise biomedical knowledge graph for robust molecular interaction prediction,
T. Ma, Y . Chen, W. Tao, D. Zheng, X. Lin, P. C.-I. Pang, Y . Liu, Y . Wang, L. Wang, B. Songet al., “Learning to denoise biomedical knowledge graph for robust molecular interaction prediction,”IEEE Transactions on Knowledge and Data Engineering, vol. 36, no. 12, pp. 8682–8694, 2024
2024
-
[15]
Local–global structure-aware geometric equivariant graph representation learning for predicting protein–ligand binding affinity,
S. Chen, H. Yi, Z. You, X. Shang, Y .-A. Huang, L. Wang, and Z. Wang, “Local–global structure-aware geometric equivariant graph representation learning for predicting protein–ligand binding affinity,” IEEE Transactions on Neural Networks and Learning Systems, 2025
2025
-
[16]
SSPPI: Cross- modality enhanced protein–protein interaction prediction from sequence and structure perspectives,
X. Bi, W. Ma, H. Jiang, W. Lu, Z. Wei, and S. Zhang, “SSPPI: Cross- modality enhanced protein–protein interaction prediction from sequence and structure perspectives,”IEEE Transactions on Neural Networks and Learning Systems, 2025
2025
-
[17]
Exploring molecular pretraining model at scale,
X. Ji, Z. Wang, Z. Gao, H. Zheng, L. Zhang, G. Ke, and W. E, “Exploring molecular pretraining model at scale,” inThe Thirty-eighth Annual Conference on Neural Information Processing Systems, 2024
2024
-
[18]
Protein language pragmatic analysis and progressive transfer learning for profiling peptide–protein interactions,
S. Chen, K. Yan, X. Li, and B. Liu, “Protein language pragmatic analysis and progressive transfer learning for profiling peptide–protein interactions,”IEEE Transactions on Neural Networks and Learning Systems, 2025
2025
-
[19]
Accurate protein– protein interaction prediction: Based on multiview heterogeneous graph autoencoders and random masking,
S. Chen, Z. Tang, L. You, and C. Y .-C. Chen, “Accurate protein– protein interaction prediction: Based on multiview heterogeneous graph autoencoders and random masking,”IEEE Transactions on Neural Networks and Learning Systems, 2025
2025
-
[20]
FusionESP: Improved enzyme–substrate pair prediction by fusing protein and chemical knowl- edge,
Z. Du, W. Fu, X. Guo, D. Caragea, and Y . Li, “FusionESP: Improved enzyme–substrate pair prediction by fusing protein and chemical knowl- edge,”Journal of Chemical Information and Modeling, vol. 65, no. 6, pp. 2806–2817, 2025
2025
-
[21]
SEFP: Structure-based enzyme function prediction,
Z. Zhang, G. Yu, Z. Deng, C. Luo, C. Cai, W. Zhang, F. Hu, K.- S. Choi, Z. Wei, L. Wanget al., “SEFP: Structure-based enzyme function prediction,”IEEE Transactions on Computational Biology and Bioinformatics, 2025
2025
-
[22]
Enzyme promiscuity: mechanism and appli- cations,
K. Hult and P. Berglund, “Enzyme promiscuity: mechanism and appli- cations,”Trends in Biotechnology, vol. 25, no. 5, pp. 231–238, 2007
2007
-
[23]
Multi-modal deep learning enables efficient and accurate annotation of enzymatic active sites,
X. Wang, X. Yin, D. Jiang, H. Zhao, Z. Wu, O. Zhang, J. Wang, Y . Li, Y . Deng, H. Liuet al., “Multi-modal deep learning enables efficient and accurate annotation of enzymatic active sites,”Nature Communications, vol. 15, no. 1, p. 7348, 2024
2024
-
[24]
VIPER: A general model for prediction of enzyme substrates,
M. J. Campbell, “VIPER: A general model for prediction of enzyme substrates,”bioRxiv, pp. 2024–06, 2024
2024
-
[25]
Using deep learning to annotate the protein universe,
M. L. Bileschi, D. Belanger, D. H. Bryant, T. Sanderson, B. Carter, D. Sculley, A. Bateman, M. A. DePristo, and L. J. Colwell, “Using deep learning to annotate the protein universe,”Nature Biotechnology, vol. 40, no. 6, pp. 932–937, 2022
2022
-
[26]
Connecting chemical and protein sequence space to predict biocatalytic reactions,
A. E. Paton, D. A. Boiko, J. C. Perkins, N. I. Cemalovic, T. Resch¨utzegger, G. Gomes, and A. R. Narayan, “Connecting chemical and protein sequence space to predict biocatalytic reactions,”Nature, vol. 646, no. 8083, pp. 108–116, 2025
2025
-
[27]
VenusX: Unlocking fine-grained functional understanding of proteins,
Y . Tan, W. Gou, B. Zhong, H. Yu, L. Hong, and B. Zhou, “VenusX: Unlocking fine-grained functional understanding of proteins,” inThe Fourteenth International Conference on Learning Representations, 2026. [Online]. Available: https://openreview.net/forum?id=zcmL592XRG
2026
-
[28]
Reactzyme: A benchmark for enzyme-reaction prediction,
C. Hua, B. Zhong, S. Luan, L. Hong, G. Wolf, D. Precup, and S. Zheng, “Reactzyme: A benchmark for enzyme-reaction prediction,”Advances in Neural Information Processing Systems, vol. 37, pp. 26 415–26 442, 2024
2024
-
[29]
A comprehensive survey of deep learning techniques in protein function prediction,
R. Dhanuka, J. P. Singh, and A. Tripathi, “A comprehensive survey of deep learning techniques in protein function prediction,”IEEE/ACM Transactions on Computational Biology and Bioinformatics, vol. 20, no. 3, pp. 2291–2301, 2023
2023
-
[30]
Protein function prediction as approximate semantic entailment,
M. Kulmanov, F. J. Guzm ´an-Vega, P. Duek Roggli, L. Lane, S. T. Arold, and R. Hoehndorf, “Protein function prediction as approximate semantic entailment,”Nature Machine Intelligence, vol. 6, no. 2, pp. 220–228, 2024
2024
-
[31]
VenusFactory: An integrated system for protein engineering with data retrieval and language model fine- tuning,
Y . Tan, C. Liu, J. Gao, W. Banghao, M. Li, R. Wang, L. Zhang, H. Yu, G. Fan, L. Honget al., “VenusFactory: An integrated system for protein engineering with data retrieval and language model fine- tuning,” inProceedings of the 63rd Annual Meeting of the Association for Computational Linguistics, 2025, pp. 230–241
2025
-
[32]
Deep learning-driven insights into enzyme–substrate interaction discovery,
W. Qian, X. Wang, Y . Huang, Y . Kang, P. Pan, C.-Y . Hsieh, and T. Hou, “Deep learning-driven insights into enzyme–substrate interaction discovery,”Journal of Chemical Information and Modeling, vol. 65, no. 1, pp. 187–200, 2024
2024
-
[33]
Z. Nie, H. Zhang, H. Jiang, Y . Liu, X. Huang, F. Xu, J. Fu, Z. Ren, Y . Tian, W.-B. Zhanget al., “OmniESI: A unified framework for enzyme-substrate interaction prediction with progressive conditional deep learning,”arXiv:2506.17963, 2025
-
[34]
Enzyme specificity prediction using cross attention graph neural networks,
H. Cui, Y . Su, T. J. Dean, T. Yu, Z. Zhang, J. Peng, D. Shukla, and H. Zhao, “Enzyme specificity prediction using cross attention graph neural networks,”Nature, pp. 1–3, 2025
2025
-
[35]
Retrieval- augmented generation for knowledge-intensive NLP tasks,
P. Lewis, E. Perez, A. Piktus, F. Petroni, V . Karpukhin, N. Goyal, H. K ¨uttler, M. Lewis, W.-t. Yih, T. Rockt ¨aschelet al., “Retrieval- augmented generation for knowledge-intensive NLP tasks,”Advances in Neural Information Processing Systems, vol. 33, pp. 9459–9474, 2020
2020
-
[36]
A survey on RAG meeting LLMs: Towards retrieval-augmented large language models,
W. Fan, Y . Ding, L. Ning, S. Wang, H. Li, D. Yin, T.-S. Chua, and Q. Li, “A survey on RAG meeting LLMs: Towards retrieval-augmented large language models,” inProceedings of the 30th ACM SIGKDD Conference on Knowledge Discovery and Data Mining, 2024, pp. 6491–6501
2024
-
[37]
Protriever: End-to-end differentiable protein homology search for fitness prediction,
R. Weitzman, P. M. Groth, L. V . Niekerk, A. Otani, Y . Gal, D. S. Marks, and P. Notin, “Protriever: End-to-end differentiable protein homology search for fitness prediction,” inForty-second International Conference on Machine Learning, 2025
2025
-
[38]
Large language models are in-context molecule learners,
J. Li, W. Liu, Z. Ding, W. Fan, Y . Li, and Q. Li, “Large language models are in-context molecule learners,”IEEE Transactions on Knowledge and Data Engineering, 2025
2025
-
[39]
Retrieval-Augmented Generation for Large Language Models: A Survey
Y . Gao, Y . Xiong, X. Gao, K. Jia, J. Pan, Y . Bi, Y . Dai, J. Sun, H. Wang, H. Wanget al., “Retrieval-augmented generation for large language models: A survey,”arXiv:2312.10997, vol. 2, no. 1, p. 32, 2023
work page internal anchor Pith review Pith/arXiv arXiv 2023
-
[40]
Graph retrieval-augmented generation: A survey,
B. Peng, Y . Zhu, Y . Liu, X. Bo, H. Shi, C. Hong, Y . Zhang, and S. Tang, “Graph retrieval-augmented generation: A survey,”ACM Transactions on Information Systems, vol. 44, no. 2, pp. 1–52, 2025
2025
-
[41]
A deep retrieval-enhanced meta-learning framework for enzyme opti- mum ph prediction,
L. Zhang, K. Luo, Z. Zhou, Y . Yu, F. Jiang, B. Wu, M. Li, and L. Hong, “A deep retrieval-enhanced meta-learning framework for enzyme opti- mum ph prediction,”Journal of Chemical Information and Modeling, vol. 65, no. 7, pp. 3761–3770, 2025. SUBMITTED TO IEEE TRANSACTIONS ON KNOWLEDGE AND DATA ENGINEERING 13
2025
-
[42]
Tranception: protein fitness prediction with au- toregressive transformers and inference-time retrieval,
P. Notin, M. Dias, J. Frazer, J. Marchena-Hurtado, A. N. Gomez, D. Marks, and Y . Gal, “Tranception: protein fitness prediction with au- toregressive transformers and inference-time retrieval,” inInternational Conference on Machine Learning. PMLR, 2022, pp. 16 990–17 017
2022
-
[43]
N. Datta, S. Shatabda, and M. S. Rahman, “Embedding is (almost) all you need: Retrieval-augmented inference for generalizable genomic prediction tasks,”arXiv:2508.04757, 2025
-
[44]
Y . Tan, R. Wang, B. Wu, L. Hong, and B. Zhou, “From high-throughput evaluation to wet-lab studies: advancing mutation effect prediction with a retrieval-enhanced model,”Bioinformatics, vol. 41, 07 2025. [Online]. Available: https://doi.org/10.1093/bioinformatics/btaf189
-
[45]
Billion-scale similarity search with GPUs,
J. Johnson, M. Douze, and H. J ´egou, “Billion-scale similarity search with GPUs,”IEEE Transactions on Big Data, vol. 7, no. 3, pp. 535– 547, 2019
2019
-
[46]
Focal loss for dense object detection,
T.-Y . Lin, P. Goyal, R. Girshick, K. He, and P. Doll ´ar, “Focal loss for dense object detection,” inProceedings of the IEEE International Conference on Computer Vision, 2017, pp. 2980–2988
2017
-
[47]
MMseqs2 enables sensitive protein sequence searching for the analysis of massive data sets,
M. Steinegger and J. S ¨oding, “MMseqs2 enables sensitive protein sequence searching for the analysis of massive data sets,”Nature Biotechnology, vol. 35, no. 11, pp. 1026–1028, 2017
2017
-
[48]
Decoupled weight decay regularization,
I. Loshchilov and F. Hutter, “Decoupled weight decay regularization,” in International Conference on Learning Representations, 2019. [Online]. Available: https://openreview.net/forum?id=Bkg6RiCqY7
2019
-
[49]
Random forests,
L. Breiman, “Random forests,”Machine Learning, vol. 45, no. 1, pp. 5–32, 2001
2001
-
[50]
LightGBM: A highly efficient gradient boosting decision tree,
G. Ke, Q. Meng, T. Finley, T. Wang, W. Chen, W. Ma, Q. Ye, and T.- Y . Liu, “LightGBM: A highly efficient gradient boosting decision tree,” Advances in Neural Information Processing Systems, vol. 30, 2017
2017
-
[51]
Evolutionary-scale prediction of atomic- level protein structure with a language model,
Z. Lin, H. Akin, R. Rao, B. Hie, Z. Zhu, W. Lu, N. Smetanin, R. Verkuil, O. Kabeli, Y . Shmueliet al., “Evolutionary-scale prediction of atomic- level protein structure with a language model,”Science, vol. 379, no. 6637, pp. 1123–1130, 2023
2023
-
[52]
Uni-mol2: Exploring molecular pretraining model at scale,
X. Ji, Z. Wang, Z. Gao, H. Zheng, L. Zhang, G. Keet al., “Uni-mol2: Exploring molecular pretraining model at scale,”arXiv:2406.14969, 2024
-
[53]
A multimodal transformer network for protein-small molecule interactions enhances predictions of kinase inhibition and enzyme-substrate relationships,
A. Kroll, S. Ranjan, and M. J. Lercher, “A multimodal transformer network for protein-small molecule interactions enhances predictions of kinase inhibition and enzyme-substrate relationships,”PLOS Computa- tional Biology, vol. 20, no. 5, p. e1012100, 2024
2024
-
[54]
KEGG: kyoto encyclopedia of genes and genomes,
M. Kanehisa and S. Goto, “KEGG: kyoto encyclopedia of genes and genomes,”Nucleic Acids Research, vol. 28, no. 1, pp. 27–30, 2000
2000
-
[55]
Autodock vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading,
O. Trott and A. J. Olson, “Autodock vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading,”Journal of Computational Chemistry, vol. 31, no. 2, pp. 455–461, 2010
2010
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.