Impact of dynamic electrostatic disorder on hole mobility in rubrene: a nonadiabatic molecular dynamics investigation
Pith reviewed 2026-06-26 23:31 UTC · model grok-4.3
The pith
Dynamic electrostatic disorder in rubrene reduces hole mobility from 35 to 21 cm² V^{-1}s^{-1} by increasing site energy disorder and shrinking the wavefunction.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
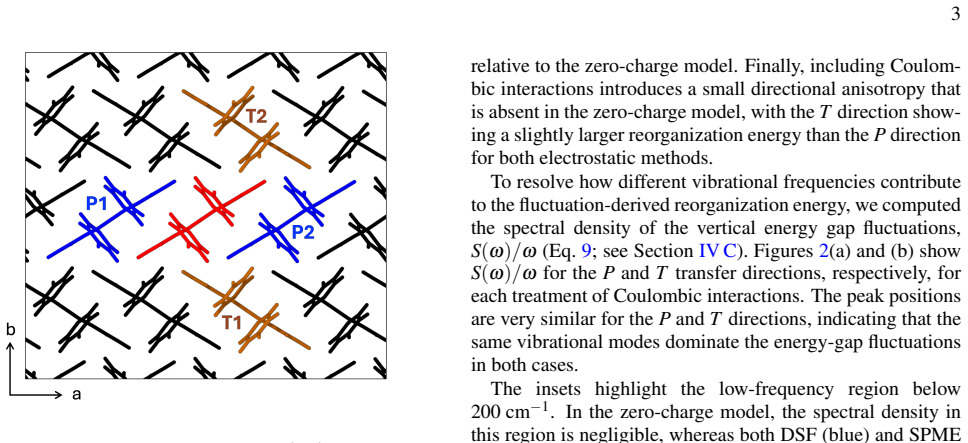
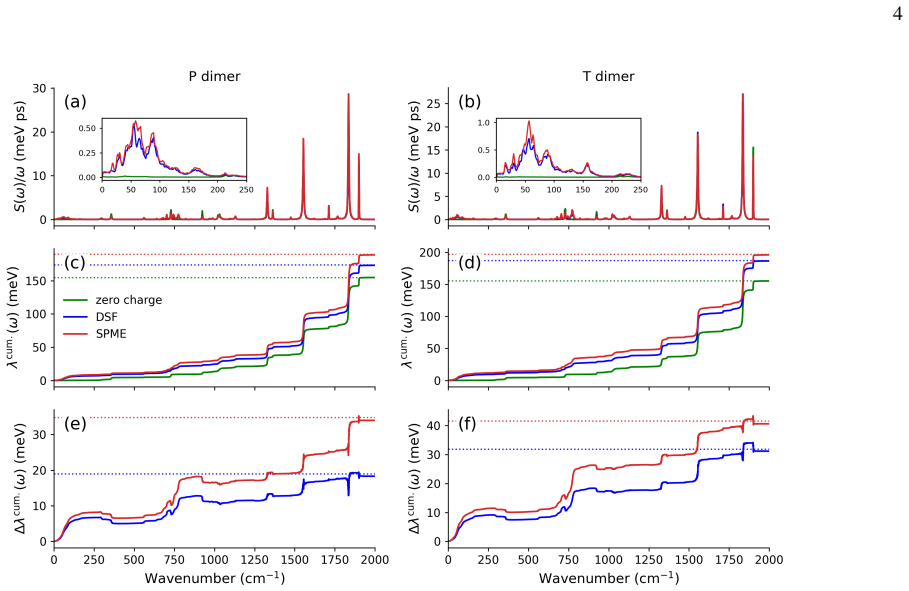
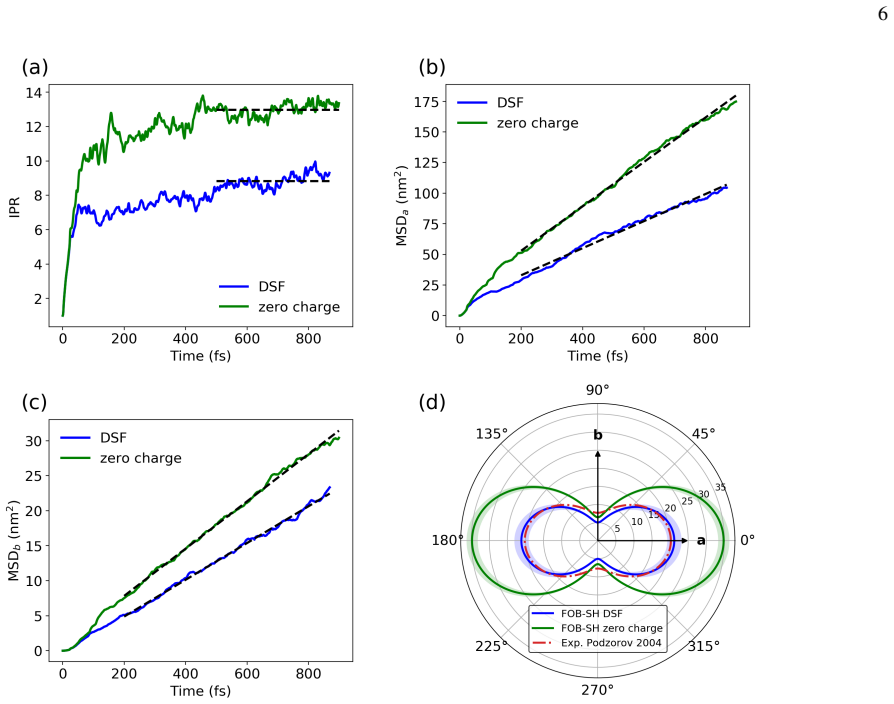
In FOB-SH simulations of room-temperature hole transport in rubrene, dynamic electrostatic disorder increases reorganization energy by 29 meV along a and 39 meV along b relative to a 152 meV baseline, raises site energy disorder, decreases the inverse participation ratio of the hole wavefunction from 13 to 9, and lowers mobility along the high-mobility direction from 35 to 21 cm² V^{-1}s^{-1}, bringing the result into close agreement with experiment.
What carries the argument
Fragment orbital-based surface hopping (FOB-SH) combined with the damped shifted-force real-space electrostatic summation method and an addition-subtraction scheme to include dynamic electrostatic disorder.
If this is right
- Electrostatic interactions raise the reorganization energy for nearest-neighbor hopping by tens of meV along both crystal axes.
- The hole wavefunction becomes more localized, as shown by the drop in inverse participation ratio.
- Mobility predictions move into agreement with measured values without any reparameterization of the underlying model.
- The same electrostatic treatment can be applied to other apolar molecular crystals previously simulated without it.
Where Pith is reading between the lines
- Simulations of similar organic crystals that omit electrostatic disorder are likely to overestimate carrier delocalization and mobility.
- The approach could be extended to electron transport or to crystals with permanent dipoles to check whether the same reduction occurs.
- Device models for organic electronics may need to incorporate dynamic electrostatic fluctuations to predict performance accurately.
Load-bearing premise
The damped shifted-force real-space electrostatic summation method combined with an addition-subtraction scheme accurately captures dynamic electrostatic disorder in fragment orbital-based surface hopping without introducing significant artifacts.
What would settle it
A room-temperature hole mobility measurement on rubrene single crystals that deviates significantly from 21 cm² V^{-1}s^{-1} when compared against an otherwise identical simulation run without the electrostatic terms would falsify the central claim.
Figures
read the original abstract
High-mobility organic molecular crystals such as rubrene are important materials for organic electronics, yet a quantitatively predictive description of their charge transport properties remains challenging. Direct mixed quantum-classical nonadiabatic molecular dynamics simulations provide a promising route by explicitly propagating the charge carrier wavefunction, without assuming a specific transport mechanism. However, previous large-scale simulations of apolar molecular crystals have commonly neglected dynamic electrostatic disorder, since evaluation of electrostatic interactions is computationally demanding and the approximation appears plausible for apolar systems. Here, we use the damped shifted-force (DSF) real-space electrostatic summation method, combined with an efficient addition-subtraction scheme, to include dynamic electrostatic disorder in fragment orbital-based surface hopping (FOB-SH) simulations of room-temperature hole transport in rubrene. We find that electrostatic interactions increase the reorganization energy for (hypothetical) nearest neighbour hopping by 29 and 39 meV along the a and b-directions, respectively, relative to a baseline value of 152 meV obtained without electrostatics. In FOB-SH simulations, electrostatic interactions lead to increased site energy disorder, reducing the spatial extent of the hole wavefunction, as measured by a decrease in the inverse participation ratio from 13 to 9, and lowering the predicted mobility along the high-mobility direction from $35$ to $21~\mathrm{cm^2 V^{-1}s^{-1}}$, in close agreement with experiment.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript reports FOB-SH nonadiabatic MD simulations of hole transport in rubrene that incorporate dynamic electrostatic disorder via the damped shifted-force (DSF) real-space summation plus an addition-subtraction scheme. Electrostatics are found to raise the reorganization energy by 29 meV (a-direction) and 39 meV (b-direction) relative to a 152 meV baseline, increase site-energy disorder, reduce the inverse participation ratio from 13 to 9, and lower the high-mobility-direction mobility from 35 to 21 cm² V⁻¹ s⁻¹, bringing it into agreement with experiment.
Significance. If the central numerical results are robust, the work supplies direct evidence that dynamic electrostatic disorder remains quantitatively important even in an apolar crystal and demonstrates an efficient route to include it in large-scale FOB-SH trajectories. The forward-simulation character (no fitted mobility or disorder parameters) and explicit wave-function propagation are strengths that distinguish the approach from phenomenological models.
major comments (1)
- [Abstract / Methods] Abstract / Methods: The headline result (IPR drop 13→9 and mobility drop 35→21 cm² V⁻¹ s⁻¹) rests on the assertion that DSF plus addition-subtraction faithfully reproduces the dynamic electrostatic site-energy fluctuations. No section compares the resulting site-energy variance or fluctuation spectrum against a reference Ewald or PME calculation performed on the same rubrene trajectories or supercells; the reorganization-energy shifts are reported only relative to the no-electrostatics baseline. This validation gap is load-bearing for the claim that the observed disorder increase is physical rather than an artifact of the electrostatic cutoff or summation scheme.
minor comments (2)
- [Abstract] Abstract: Reported mobility and IPR values are given without error bars, standard deviations across trajectories, or convergence tests with respect to supercell size or time step.
- [Abstract] Abstract: The baseline reorganization energy of 152 meV is stated without indicating whether it was obtained from the same FOB-SH trajectories or from a separate calculation, and without quoting its statistical uncertainty.
Simulated Author's Rebuttal
We thank the referee for their positive assessment of the work's significance and for the constructive comment on validation. We respond point-by-point below.
read point-by-point responses
-
Referee: [Abstract / Methods] Abstract / Methods: The headline result (IPR drop 13→9 and mobility drop 35→21 cm² V⁻¹ s⁻¹) rests on the assertion that DSF plus addition-subtraction faithfully reproduces the dynamic electrostatic site-energy fluctuations. No section compares the resulting site-energy variance or fluctuation spectrum against a reference Ewald or PME calculation performed on the same rubrene trajectories or supercells; the reorganization-energy shifts are reported only relative to the no-electrostatics baseline. This validation gap is load-bearing for the claim that the observed disorder increase is physical rather than an artifact of the electrostatic cutoff or summation scheme.
Authors: We acknowledge that the manuscript does not contain a direct side-by-side comparison of site-energy variance or fluctuation spectra from DSF+addition-subtraction versus Ewald/PME on identical rubrene trajectories. The DSF approach with the addition-subtraction correction was selected for computational tractability in large-scale FOB-SH runs; its accuracy for electrostatic potentials in periodic molecular systems is documented in the cited methodological literature. Reorganization energies are obtained self-consistently under the same electrostatic treatment for charged and neutral states, and the reported increase (29–39 meV) is therefore an internal difference attributable to the explicit inclusion of dynamic electrostatics. We maintain that the physical origin of the increased disorder and reduced IPR/mobility is robust within this consistent framework. No new Ewald calculations on the full trajectories are planned, as they would be prohibitive, but a short paragraph referencing prior DSF validation benchmarks can be added if requested. revision: partial
Circularity Check
No significant circularity; forward simulation using established methods
full rationale
The paper performs explicit FOB-SH nonadiabatic molecular dynamics on rubrene trajectories, computing site energies, reorganization energies, IPR, and mobility as direct simulation outputs when electrostatic disorder is added via the DSF summation. These quantities are not defined in terms of each other, not obtained by fitting a parameter to a subset and relabeling it a prediction, and not justified solely by self-citation chains. The methodological choice of DSF plus addition-subtraction is an independent modeling decision whose accuracy can be (and is expected to be) checked against external references such as Ewald summation; it does not reduce the reported mobility or IPR values to the input assumptions by construction. The derivation chain therefore remains self-contained and non-circular.
Axiom & Free-Parameter Ledger
free parameters (1)
- baseline reorganization energy =
152 meV
axioms (2)
- domain assumption Fragment orbital-based surface hopping accurately models hole transport in molecular crystals
- domain assumption Damped shifted-force summation plus addition-subtraction scheme correctly represents dynamic electrostatic disorder
Reference graph
Works this paper leans on
-
[1]
Quantum localization and delocalization of charge carriers in organic semiconducting crystals , author=. Nat. Commun. , volume=. 2019 , publisher=
2019
-
[2]
Flickering Polarons Extending over Ten Nanometres Mediate Charge Transport in High-Mobility Organic Crystals , author=. Adv. Theory Simul. , volume=. 2020 , publisher=
2020
-
[3]
Charge transport in organic semiconductors: the perspective from nonadiabatic molecular dynamics , author=. Acc. Chem. Res. , volume=. 2022 , publisher=
2022
-
[4]
Transiently delocalized states enhance hole mobility in organic molecular semiconductors , author=. Nat. Mater. , volume=. 2023 , publisher=
2023
-
[5]
Exact method for the simulation of Coulombic systems by spherically truncated, pairwise r- 1 summation , author=. J. Chem. Phys. , volume=. 1999 , publisher=
1999
-
[6]
Is the Ewald summation still necessary? Pairwise alternatives to the accepted standard for long-range electrostatics , author=. J. Chem. Phys. , volume=. 2006 , publisher=
2006
-
[7]
Efficient Calculation of Electrostatic Energies for Large-Scale Nonadiabatic Molecular Dynamics in a Site Basis , author=. J. Chem. Theory Comput. , volume=. 2025 , publisher=
2025
-
[8]
Mechanoelectric response of single-crystal rubrene from Ab initio molecular dynamics , author=. J. Phys. Chem. Lett. , volume=. 2021 , publisher=
2021
-
[9]
Thermoelectric transport in molecular crystals driven by gradients of thermal electronic disorder , author=. Sci. Adv. , volume=. 2024 , publisher=
2024
-
[10]
Long spin lifetimes of charge carriers in rubrene crystals due to fast transient-localization motion , author=. Nat. Commun. , volume=. 2025 , publisher=
2025
-
[11]
A new frontier in exciton transport: transient delocalization , author=. J. Phys. Chem. Lett. , volume=. 2022 , publisher=
2022
-
[12]
Exciton transport in molecular organic semiconductors boosted by transient quantum delocalization , author=. Nat. Commun. , volume=. 2022 , publisher=
2022
-
[13]
Transiently delocalised hybrid quantum states are gateways for efficient exciton dissociation at organic donor-acceptor interfaces , author=. Nat. Commun. , volume=. 2025 , publisher=
2025
-
[14]
Transient localization in crystalline organic semiconductors , author=. Phys. Rev. B , volume=. 2011 , publisher=
2011
-
[15]
The transient localization scenario for charge transport in crystalline organic materials , author=. Adv. Funct. Mater. , volume=. 2016 , publisher=
2016
-
[16]
Intrinsic charge transport on the surface of organic semiconductors , author=. Phys. Rev. Lett. , volume=. 2004 , publisher=
2004
-
[17]
Tunable Fr
Hulea, Iulian N and Fratini, Simone and Xie, H and Mulder, Cornelis L and Iossad, Nikolai N and Rastelli, Gianluca and Ciuchi, Sergio and Morpurgo, Alberto F , journal=. Tunable Fr. 2006 , publisher=
2006
-
[18]
High-mobility transistors based on single crystals of isotopically substituted rubrene-d 28 , author=. J. Phys. Chem. C , volume=. 2013 , publisher=
2013
-
[19]
Molecular dynamics with electronic transitions , author=. J. Chem. Phys. , volume=. 1990 , publisher=
1990
-
[20]
Ultrafast electronic coupling estimators: Neural networks versus physics-based approaches , author=. J. Chem. Theory Comput. , volume=. 2023 , publisher=
2023
-
[21]
Recent advances in the theory and molecular simulation of biological electron transfer reactions , author=. Chem. Rev. , volume=. 2015 , publisher=
2015
-
[22]
Quantum versus classical electron transfer energy as reaction coordinate for the aqueous Ru2+/Ru3+ redox reaction , author=. Theor. Chem. Acc. , volume=. 2006 , publisher=
2006
-
[23]
FOB-SH: Fragment orbital-based surface hopping for charge carrier transport in organic and biological molecules and materials , author=. J. Chem. Phys. , volume=. 2016 , publisher=
2016
-
[24]
How to calculate charge mobility in molecular materials from surface hopping non-adiabatic molecular dynamics--beyond the hopping/band paradigm , author=. Phys. Chem. Chem. Phys. , volume=. 2019 , publisher=
2019
-
[25]
Ultrafast estimation of electronic couplings for electron transfer between -conjugated organic molecules , author=. J. Chem. Theory Comput. , volume=. 2014 , publisher=
2014
-
[26]
II , author=
Ultrafast estimation of electronic couplings for electron transfer between pi-conjugated organic molecules. II , author=. J. Chem. Phys. , volume=. 2021 , publisher=
2021
-
[27]
Detailed balance, internal consistency, and energy conservation in fragment orbital-based surface hopping , author=. J. Chem. Phys. , volume=. 2017 , publisher=
2017
-
[28]
Electronic couplings for charge transfer across molecule/metal and molecule/semiconductor interfaces: Performance of the projector operator-based diabatization approach , author=. J. Phys. Chem. C. , volume=. 2017 , publisher=
2017
-
[29]
Development and testing of a general amber force field , author=. J. Comput. Chem. , volume=. 2004 , publisher=
2004
-
[30]
Charge transport in high-mobility conjugated polymers and molecular semiconductors , author=. Nat. Mater. , volume=. 2020 , publisher=
2020
-
[31]
A smooth particle mesh Ewald method , author=. J. Chem. Phys. , volume=. 1995 , publisher=
1995
-
[32]
Small Methods , volume=
Organic flexible electronics , author=. Small Methods , volume=. 2018 , publisher=
2018
-
[33]
Organic semiconductors and their applications in photovoltaic devices , author=. Polym. Rev. , volume=. 2012 , publisher=
2012
-
[34]
Prediction of the absolute charge mobility of molecular semiconductors: the case of rubrene , author=. Adv. Mater. , volume=. 2007 , publisher=
2007
-
[35]
Transport properties in the rubrene crystal: electronic coupling and vibrational reorganization energy , author=. Adv. Mater. , volume=. 2005 , publisher=
2005
-
[36]
Troisi, Alessandro , doi =. J. Chem. Phys. , mendeley-groups =
-
[37]
Vehoff, Thorsten and Baumeier, Bj. J. Am. Chem. Soc. , mendeley-groups =. doi:10.1021/ja104380c , file =
-
[38]
Nan, Guangjun and Yang, Xiaodi and Wang, Linjun and Shuai, Zhigang and Zhao, Yi , doi =. Phys. Rev. B , mendeley-groups =
-
[39]
Jiang, Yuqian and Zhong, Xinxin and Shi, Wen and Peng, Qian and Geng, Hua and Zhao, Yi and Shuai, Zhigang , doi =. Nanoscale Horiz. , mendeley-groups =. arXiv , arxivId =:arXiv:1408.1149 , file =
-
[40]
Li, Weitang and Ren, Jiajun and Shuai, Zhigang , doi =. J. Phys. Chem. Lett. , mendeley-groups =
-
[41]
Phys. Rev. Lett. , mendeley-groups =. doi:10.1103/PhysRevLett.114.086601 , file =
-
[42]
Runeson, Johan E. and Drayton, Thomas J.G. and Manolopoulos, David E. , doi =. J. Chem. Phys. , mendeley-groups =. arXiv , arxivId =:2406.19851 , file =
-
[43]
Crossover from incoherent to coherent electron tunneling between defects in MgO , author=. Phys. Rev. B , volume=. 2012 , publisher=
2012
-
[44]
and Jang, Wooik and Hambsch, Mike and Mannsfeld, Stefan C
Wang, Zichen and Buchholtz, Stephanie A. and Jang, Wooik and Hambsch, Mike and Mannsfeld, Stefan C. B. and Ren, Xinglong and Jacobs, Ian E. and Sirringhaus, Henning and Kleemann, Hans , doi =. ACS Nano , keywords =
-
[45]
Sakamoto, Yuji and Izawa, Seiichiro and Ohkita, Hideo and Hiramoto, Masahiro and Tamai, Yasunari , doi =. Commun. Mater. , mendeley-groups =
-
[46]
Izawa, Seiichiro and Hiramoto, Masahiro , doi =. Nat. Photonics , mendeley-groups =
-
[47]
An approach to computing electrostatic charges for molecules , author=. J. Comput. Chem. , volume=. 1984 , publisher=
1984
-
[48]
Semiclassical trajectory studies of electron-transfer and proton-transfer reactions , author=
Dynamics of reactions in polar solvents. Semiclassical trajectory studies of electron-transfer and proton-transfer reactions , author=. J. Phys. Chem. , volume=. 1982 , publisher=
1982
-
[49]
Effects of solvent and solute polarizability on the reorganization energy of electron transfer , author=. J. Phys. Chem. A , volume=. 2004 , publisher=
2004
-
[50]
High mobility emissive organic semiconductors for optoelectronic devices , author=. J. Am. Chem. Soc. , volume=. 2025 , publisher=
2025
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.