First-Principles Insights into Surface and Ligand Effects in Stoichiometric HgTe Quantum Dots
Pith reviewed 2026-06-27 18:29 UTC · model grok-4.3
The pith
Neutral ligands eliminate surface-derived localized states in 1.8 nm stoichiometric HgTe nanoclusters by restoring coordination and hybridizing band edges.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
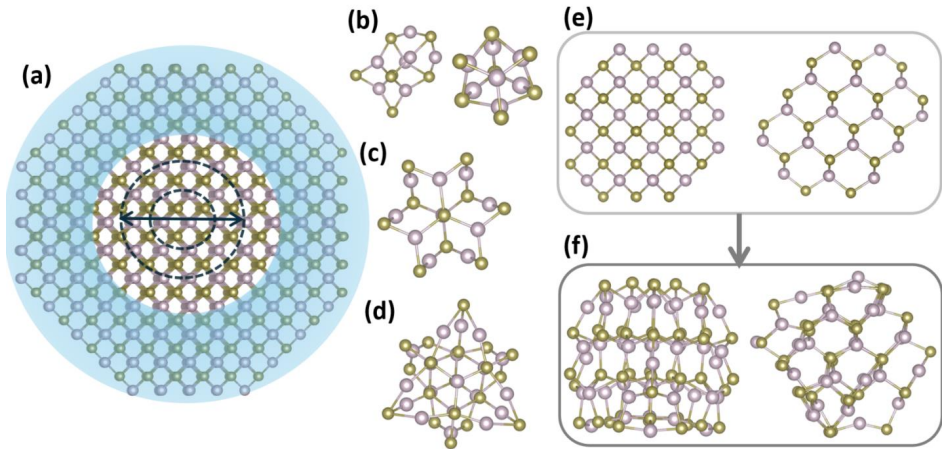
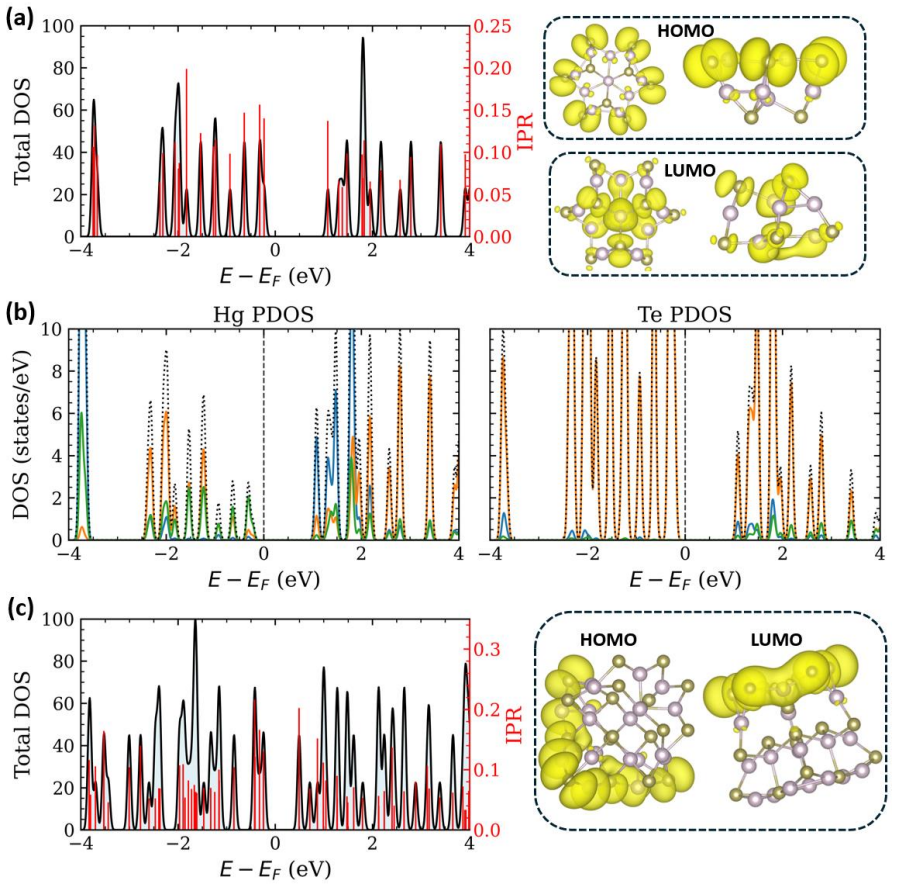
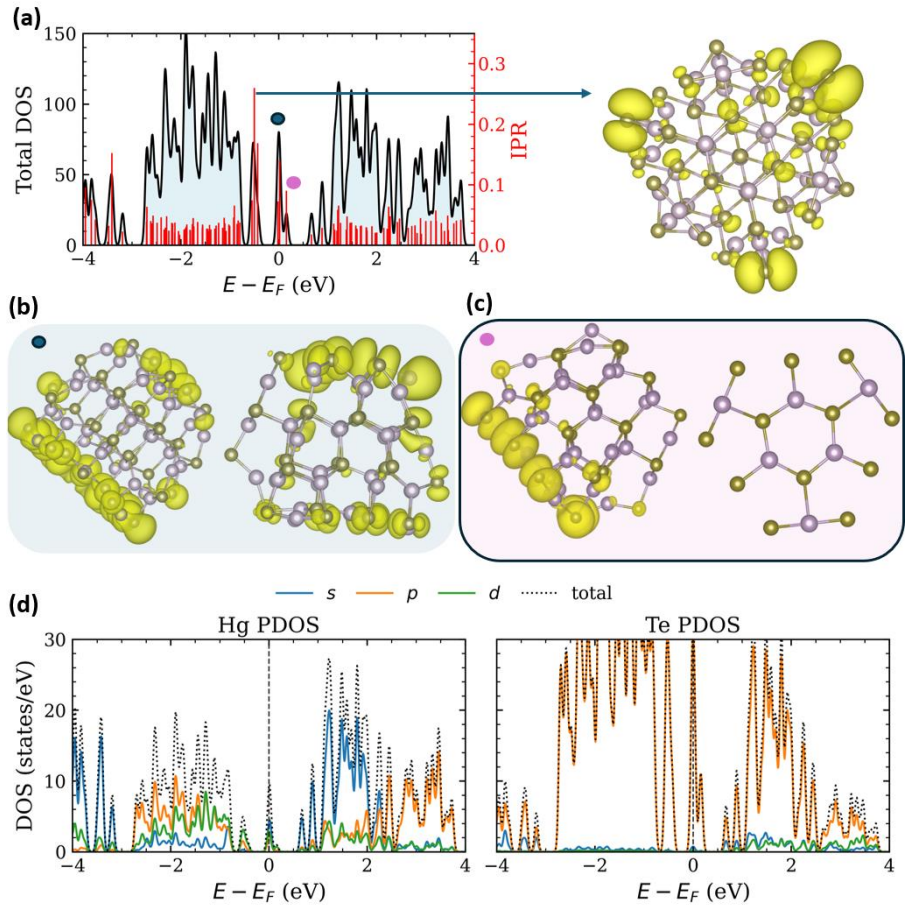
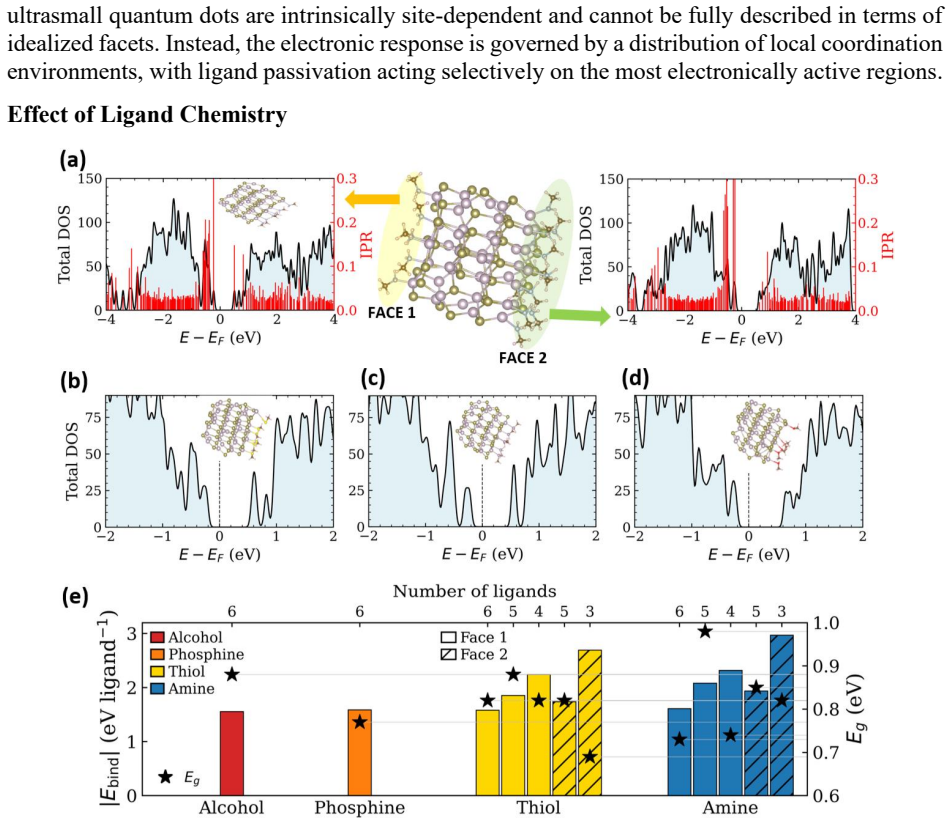
Stoichiometric HgTe nanoclusters from 0.86 to 1.85 nm transition from confinement-dominated delocalized states in small clusters to surface-influenced localized near-gap states in larger clusters due to undercoordination and bond inhomogeneity; neutral ligands such as amines, thiols, phosphines, and alcohols eliminate these surface-derived states in 1.8 nm clusters by restoring coordination and hybridizing the band-edge structure, with bandgap sensitivity to ligand identity and site.
What carries the argument
Size-dependent stoichiometric HgTe cluster models that track how surface undercoordination produces localized states and how neutral ligand attachment restores coordination while hybridizing frontier orbitals.
If this is right
- Band edge states spatially separate at intermediate sizes without creating deep gap states.
- Ligand choice and binding location directly tune the bandgap.
- Neutral ligands function as chemical controls for frontier state engineering in infrared optoelectronics.
- Surface coordination and ligand chemistry together determine the electronic structure.
Where Pith is reading between the lines
- Ligand selection may permit electronic tuning at fixed cluster size for device optimization.
- The surface passivation mechanism could generalize to other small mercury chalcogenide clusters.
- Synthesis targeting specific ligand sites on 1.8 nm clusters would test the predicted hybridization effects.
Load-bearing premise
The chosen simulation approach and stoichiometric cluster models correctly capture the real electronic structure and surface chemistry of HgTe nanoclusters.
What would settle it
Detection of persistent localized surface states or large mismatches between simulated and measured bandgaps in experimentally prepared 1.8 nm HgTe clusters passivated with the neutral ligands examined here.
Figures

read the original abstract
HgTe quantum dots are promising mid-infrared nanomaterials owing to their exceptional bandgap tunability, yet their electronic structure is strongly influenced by surface coordination and ligand passivation at ultrasmall sizes. Here, we employ atomistic simulations to systematically investigate stoichiometric HgTe nanoclusters with sizes 0.86 to 1.85 nm. The in silico exploration uncovers a transition from confinement-dominated electronic structures with delocalized frontier states in small self-passivated clusters to surface influenced characteristics in larger nanoclusters. Increased coordination and bond-length inhomogeneity in the larger nanoclusters generate localized near-gap states centered on undercoordinated surface atoms. At intermediate sizes, the band edge states become spatially separated on different regions of the cluster without forming deep gap states, marking the onset of surface induced electronic asymmetry. In larger clusters (1.8 nm), common neutral ligands like amines, thiols, phosphines, and alcohols effectively eliminate surface-derived localized states by restoring local coordination and altering the band edge electronic structure through ligand surface hybridization. The sensitivity of the bandgap to ligand identity and binding site underscores the interplay between surface coordination and ligand chemistry in shaping the electronic structure of these nanoclusters. These insights provide an atomistic understanding of size-dependent electronic structures in ultrasmall HgTe clusters. The study further establishes neutral ligands as powerful chemical handles for engineering frontier electronic states relevant to infrared optoelectronic functionality.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript reports atomistic simulations of stoichiometric HgTe nanoclusters (0.86–1.85 nm) that identify a size-dependent transition from confinement-dominated delocalized frontier states in small self-passivated clusters to surface-influenced localized near-gap states in larger clusters; it further claims that common neutral ligands (amines, thiols, phosphines, alcohols) in 1.8 nm clusters eliminate these surface-derived states by restoring local coordination and inducing ligand-surface hybridization, with bandgap sensitivity to ligand identity.
Significance. If the computational results hold, the work supplies atomistic understanding of how surface coordination and ligand chemistry shape the electronic structure of ultrasmall HgTe quantum dots, identifying neutral ligands as handles for engineering band-edge states relevant to mid-infrared optoelectronics.
major comments (2)
- Abstract: the central claim that neutral ligands eliminate surface-derived localized states in 1.8 nm clusters via restored coordination and hybridization rests on unshown simulation details; no functional, basis set, relativistic treatment, convergence tests, or error bars are supplied, rendering the mapping from computed DOS to the claimed mechanism impossible to evaluate.
- The interpretation of ligand effects assumes the chosen atomistic method (presumed DFT) accurately locates and removes surface states without systematic errors from functional choice or neglected many-body renormalization of the narrow-gap HgTe structure; no benchmarks against experiment or higher-level theory are reported to support this.
Simulated Author's Rebuttal
We thank the referee for their constructive and detailed comments, which have helped us improve the clarity and completeness of the manuscript. We provide point-by-point responses below and have revised the manuscript to address the concerns where possible.
read point-by-point responses
-
Referee: Abstract: the central claim that neutral ligands eliminate surface-derived localized states in 1.8 nm clusters via restored coordination and hybridization rests on unshown simulation details; no functional, basis set, relativistic treatment, convergence tests, or error bars are supplied, rendering the mapping from computed DOS to the claimed mechanism impossible to evaluate.
Authors: We agree that the abstract is a high-level summary and does not contain methodological specifics. The full details—including the PBE functional, def2-TZVP basis set, ZORA relativistic treatment, SCF and geometry convergence thresholds, and error estimation from multiple initial configurations—are provided in the Methods section and Supplementary Information. To improve accessibility, we have revised the abstract to include a concise statement of the computational approach and have added error bars to the relevant DOS and bandgap data in the figures and main text. revision: yes
-
Referee: The interpretation of ligand effects assumes the chosen atomistic method (presumed DFT) accurately locates and removes surface states without systematic errors from functional choice or neglected many-body renormalization of the narrow-gap HgTe structure; no benchmarks against experiment or higher-level theory are reported to support this.
Authors: We acknowledge that standard DFT can introduce systematic errors in narrow-gap semiconductors and does not capture many-body renormalization. In the revised manuscript we have expanded the discussion to justify the PBE choice with references to its performance on related II-VI clusters, explicitly note the qualitative character of the ligand-hybridization trends, and outline the expected direction of GW corrections. Direct benchmarks against experiment or higher-level theory for these specific ligand-passivated clusters are not included in the present work; such calculations lie beyond the current scope but are identified as important future directions. revision: partial
Circularity Check
No circularity: results are direct outputs of atomistic simulations
full rationale
The paper reports size-dependent electronic structure trends and ligand effects in HgTe nanoclusters exclusively as outputs of atomistic simulations (DFT-based cluster calculations). No equations, fitted parameters, or self-citations are presented that reduce any claim to an input by construction, nor are uniqueness theorems or ansatzes imported from prior author work. The central claims rest on computed densities of states, coordination numbers, and hybridization patterns obtained from the models themselves, making the derivation chain self-contained against external benchmarks.
Axiom & Free-Parameter Ledger
axioms (1)
- domain assumption Standard density-functional-theory approximations suffice to capture qualitative trends in electronic structure and surface states of stoichiometric HgTe clusters.
Reference graph
Works this paper leans on
-
[2]
(16) Giansante, C.; Infante, I
https://doi.org/10.1021/acs.chemmater.6b04648. (16) Giansante, C.; Infante, I. Surface Traps in Colloidal Quantum Dots: A Combined Experimental and Theoretical Perspective. J. Phys. Chem. Lett. 2017, 8 (20), 5209–5215. https://doi.org/10.1021/acs.jpclett.7b02193. (17) Coffey, B.; Skytte, E.; Ahmed, T.; Vasileiadou, E. S.; Lin, E. Y .; Sueh Hua, A.; Cook, ...
-
[3]
Hybrid Functionals Based on a Screened Coulomb Potential
https://doi.org/10.1021/jp810292n. (46) Heyd, J.; Scuseria, G. E.; Ernzerhof, M. Hybrid Functionals Based on a Screened Coulomb Potential. J. Chem. Phys. 2003, 118 (18), 8207–8215. https://doi.org/10.1063/1.1564060. (47) Heyd, J.; Scuseria, G. E.; Ernzerhof, M. Erratum: “Hybrid Functionals Based on a Screened Coulomb Potential” [J. Chem. Phys. 118, 8207 (...
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.