Exploring Multi-Transition-Metal NASICON Frameworks as High-Performance Cathodes for Sodium-Ion Batteries
Pith reviewed 2026-06-29 23:27 UTC · model grok-4.3
The pith
Calculations identify Na_x MnFe0.5Cr0.5(PO4)3 as a balanced ternary NASICON composition for sodium-ion battery cathodes.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
Among the nine NASICON frameworks examined, Na_x MnFe0.5Cr0.5(PO4)3 is singled out as the composition that simultaneously satisfies phase stability at relevant sodium contents, useful intercalation voltages dominated by Fe redox, thermodynamic (meta)stability, and low Na+ migration barriers, making it the strongest candidate for experimental follow-up among the multi-transition-metal variants.
What carries the argument
Density functional theory survey of phase behavior, average intercalation voltages, electronic band gaps, and nudged-elastic-band Na+ migration barriers across unary, binary, and ternary Mn-Fe-Cr NASICON compositions.
If this is right
- Mixed transition-metal NASICONs produce more complex Na intercalation phase behavior than their unary counterparts.
- Fe4+/Fe3+ redox activity dominates and elevates average voltages to approximately 4.0 V across the compositions.
- Frameworks containing multiple transition metals achieve the lowest Na+ migration barriers in the 0.3-0.4 eV range.
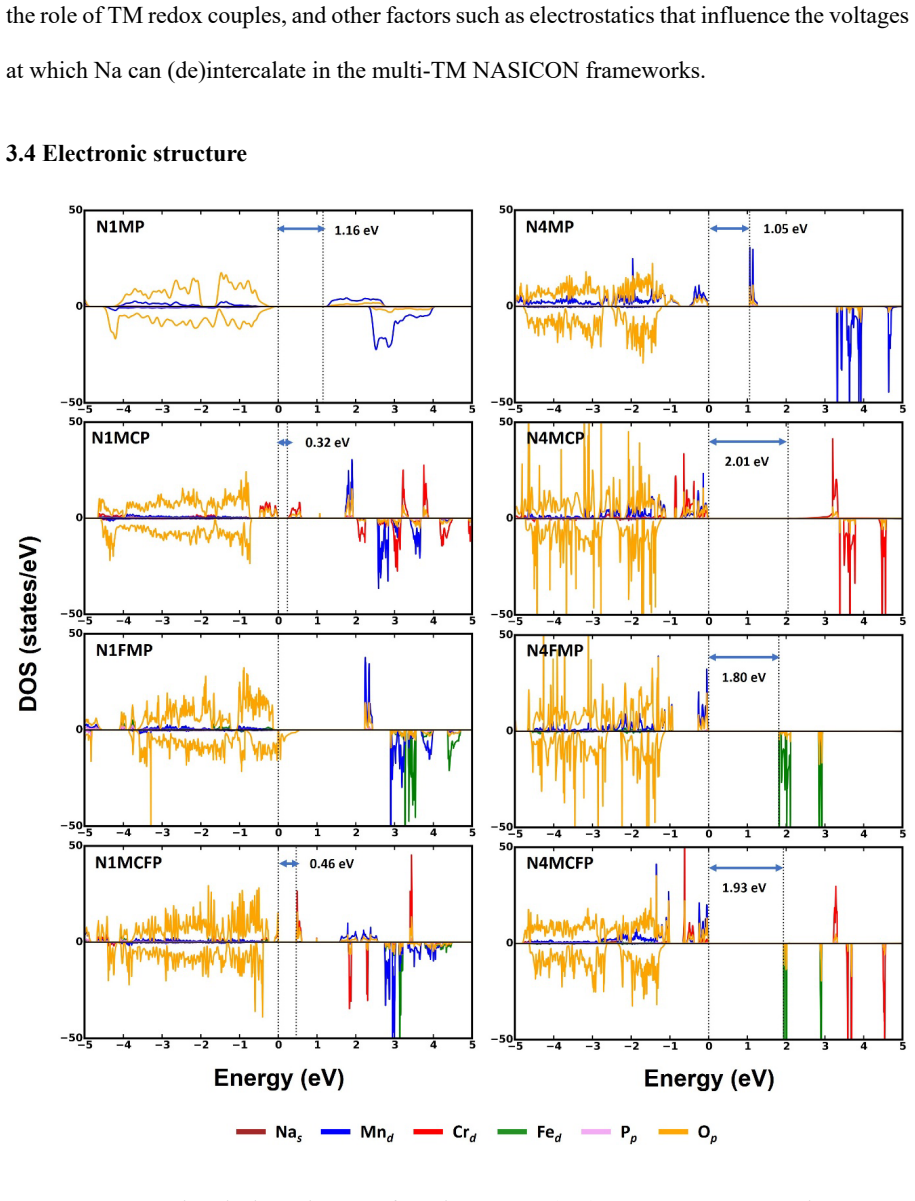
- Electronic band gaps change non-systematically when more than one transition metal is present.
Where Pith is reading between the lines
- The computational ranking supplies a short list that experimental groups can prioritize for synthesis and full-cell testing.
- The observed benefit of mixed transition metals on ion mobility may generalize to other polyanionic sodium cathode families.
- Adjusting the exact Mn:Fe:Cr ratio around the 1:0.5:0.5 composition could further tune voltage or stability if the current ternary is synthesized.
Load-bearing premise
Density functional theory calculations give accurate enough predictions of phase stability, voltages, and migration barriers for these NASICONs that no experimental correction or validation is required to rank compositions.
What would settle it
Laboratory synthesis and electrochemical cycling of Na_x MnFe0.5Cr0.5(PO4)3 that yields measured Na+ migration barriers above 0.5 eV or average voltages below 3.5 V would contradict the claim that this ternary composition is optimal.
Figures
read the original abstract
The search for sustainable, high-performance cathodes has driven a growing interest in sodium superionic conductor (NASICON)-type phosphates for sodium-ion batteries (SIBs). To identify promising NASICONs containing earth-abundant transition metals (TMs) and to systematically examine the role of multiple TMs in influencing the various properties of NASICON cathodes, we employ density functional theory calculations to investigate nine NASICON compositions containing Mn, Cr, and/or Fe, and spanning unary, binary, and ternary combinations. Our calculations reveal that unary systems, in terms of their Na intercalation phase behavior, exhibit well-defined stabilization at intermediate Na contents ($x$ in Na$_x$TM$_2$(PO$_4$)$_3$), while binary and ternary systems display more complex phase behavior, with some systems showing a shift of thermodynamic minima from $x$ = 3 to 2. Intercalation voltages highlight the dominant role of Fe$^{4+}$/Fe$^{3+}$ redox activity in elevating average voltages ($\sim$4.0 V), while Mn and Cr introduce intermediate-to-low voltage redox activity. Electronic structure data demonstrate non-systematic changes in the band gap, especially in systems containing multiple TMs. Na$^+$ mobility results identify mixed-TM frameworks as favorable, achieving Na$^+$ migration barriers in the 0.3-0.4 eV range. Importantly, we identify Na$_x$MnFe$_{0.5}$Cr$_{0.5}$(PO$_4$)$_3$ to be a promising ternary composition for subsequent experimental validation, offering an optimal intersection of phase stability, voltages, thermodynamic (meta)stability, and Na$^+$ migration barriers. Together, our study provides fundamental insights into the interplay between compositional complexity, thermodynamic stability, electronic structure, and ionic transport in NASICONs, and offers actionable design principles for utilising multi-TM NASICONs as high performance SIB cathodes.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript uses density functional theory to examine nine NASICON compositions (unary, binary, and ternary) based on Mn, Fe, and Cr, focusing on Na intercalation phase stability, voltages, electronic band gaps, thermodynamic (meta)stability, and Na+ migration barriers. It concludes that the ternary composition Na_x MnFe_{0.5}Cr_{0.5}(PO_4)_3 offers an optimal balance of these properties and is recommended for experimental validation, while providing design principles for multi-transition-metal NASICON cathodes.
Significance. If the computational results hold, the work supplies concrete, composition-specific guidance on how mixing earth-abundant transition metals modulates voltage, stability, and transport in NASICON frameworks, which could usefully inform targeted synthesis efforts in sodium-ion battery research.
major comments (1)
- [Methods] Methods section: the manuscript provides no information on the exchange-correlation functional, Hubbard U corrections (if any), plane-wave cutoff energy, k-point sampling, or convergence thresholds employed for the total-energy, voltage, and NEB calculations. Because the central recommendation of Na_x MnFe_{0.5}Cr_{0.5}(PO_4)_3 rests directly on the numerical values of voltages (~4.0 V), barriers (0.3–0.4 eV), and relative stabilities, the absence of these parameters prevents assessment of reproducibility and systematic error.
minor comments (2)
- [Abstract] Abstract, line on ternary composition: the phrase 'optimal intersection' is used without a quantitative figure of merit; a short statement of how the four criteria were weighted or ranked would improve clarity.
- Throughout: the notation Na_x TM_2(PO_4)_3 is introduced but the precise stoichiometry for the ternary case (MnFe_{0.5}Cr_{0.5}) is written inconsistently with subscripts; uniform LaTeX formatting would aid readability.
Simulated Author's Rebuttal
We thank the referee for the detailed review and constructive comment on the methods. We agree that the computational parameters must be explicitly stated to support reproducibility and assessment of the reported voltages, barriers, and stabilities. We will revise the manuscript to address this.
read point-by-point responses
-
Referee: [Methods] Methods section: the manuscript provides no information on the exchange-correlation functional, Hubbard U corrections (if any), plane-wave cutoff energy, k-point sampling, or convergence thresholds employed for the total-energy, voltage, and NEB calculations. Because the central recommendation of Na_x MnFe_{0.5}Cr_{0.5}(PO_4)_3 rests directly on the numerical values of voltages (~4.0 V), barriers (0.3–0.4 eV), and relative stabilities, the absence of these parameters prevents assessment of reproducibility and systematic error.
Authors: We agree that these details are essential and were omitted from the submitted manuscript. In the revised version we will add a dedicated Methods section that reports the exchange-correlation functional, any Hubbard U corrections (including the specific U values applied to Mn, Fe, and Cr), plane-wave cutoff energy, k-point sampling, and all convergence thresholds used for total-energy, voltage, and NEB calculations. This addition will directly enable reproducibility checks and evaluation of systematic errors in the reported ~4 V voltages and 0.3–0.4 eV barriers. revision: yes
Circularity Check
No significant circularity
full rationale
The paper's central results derive from direct DFT computations of phase stability, intercalation voltages, band gaps, and Na+ migration barriers across nine specified NASICON compositions. The recommendation of Na_x MnFe0.5Cr0.5(PO4)3 follows from comparing these independent outputs against stated criteria; no parameter is fitted to the target composition, no equation reduces to its own inputs by construction, and no load-bearing premise rests on self-citation. The work explicitly positions its outputs as inputs for future experiments rather than self-verifying predictions.
Axiom & Free-Parameter Ledger
axioms (1)
- domain assumption Standard density functional theory approximations accurately predict thermodynamic stability, redox voltages, electronic structure, and ionic migration barriers in these phosphate frameworks.
Reference graph
Works this paper leans on
-
[1]
Specifically, we examined their structural stability, phase behavior, intercalation voltages, electronic structures, and Na+ Em across unary, binary, and ternary systems
Conclusion We systematically investigated nine NASICON compositions containing earth-abundant Mn, Cr, and/or Fe using DFT calculations as potential SIB cathodes. Specifically, we examined their structural stability, phase behavior, intercalation voltages, electronic structures, and Na+ Em across unary, binary, and ternary systems. In terms of phase behavi...
2021
-
[2]
A foundation model for atomistic materials chemistry
8 D. Larcher and J.-M. Tarascon, Nature Chem, 2015, 7, 19–29. 9 J.-Y. Hwang, S.-T. Myung and Y.-K. Sun, Chem. Soc. Rev., 2017, 46, 3529–3614. 10 N. Tapia-Ruiz, A. R. Armstrong, H. Alptekin, M. A. Amores, H. Au, J. Barker, R. Boston, W. R. Brant, J. M. Brittain, Y. Chen, M. Chhowalla, Y.-S. Choi, S. I. R. Costa, M. Crespo Ribadeneyra, S. A. Cussen, E. J. C...
work page internal anchor Pith review Pith/arXiv arXiv doi:10.48550/arxiv.2401.00096 2015
-
[3]
65 M. M. Yatskin, N. Yu. Strutynska, V. N. Baumer, I. V. Ogorodnyk and N. S. Slobodyanik, Acta Crystallogr E Struct Rep Online, 2012, 68, i55–i55. 66 Z. Wang, C. Tang, Z. Wang, Q. Zhang, P. Lv, K. Yu, J. Zheng and W. Wei, Energy Mater Adv, 2023, 4,
2012
-
[4]
Harbaoui, M
67 D. Harbaoui, M. M. S. Sanad, C. Rossignol, E. K. Hlil, N. Amdouni, K. Zaidat and S. Obbade, Journal of Alloys and Compounds, 2022, 901, 163641. 68 N. El Hoda Bouftila, H. Aziam, A. Chouiekh, A. Rjeb, T. Lamcharfi, A. Faik, I. Saadoune, Y. Ababou and M. Naji, Journal of Alloys and Compounds, 2023, 961, 171054. 32 69 H. Kabbour, D. Coillot, M. Colmont, C...
2022
-
[5]
82 W. Sun, S. T. Dacek, S. P. Ong, G. Hautier, A. Jain, W. D. Richards, A. C. Gamst, K. A. Persson and G. Ceder, Sci. Adv., 2016, 2, e1600225. 83 D. B. Tekliye, A. Kumar, X. Weihang, T. D. Mercy, P. Canepa and G. Sai Gautam, Chem. Mater., 2022, 34, 10133–10143. 84 L. Vijayan, R. Cheruku, G. Govindaraj and S. Rajagopan, Materials Chemistry and Physics, 201...
2016
-
[6]
Gokulnath, S
90 S. Gokulnath, S. Krishnan, V. Yadav and M. Sathish, Energy Fuels, 2024, 38, 13407–13415. 91 Z. Rong, R. Malik, P. Canepa, G. Sai Gautam, M. Liu, A. Jain, K. Persson and G. Ceder, Chem. Mater., 2015, 27, 6016–6021. 92 R. Devi, A. Balasubramanian, K. T. Butler and G. Sai Gautam, Sci Data, 2025, 12,
2024
-
[7]
Plessow, J
93 P. Plessow, J. Chem. Theory Comput., 2013, 9, 1305–1310. 94 Q. Zhang, H. Yan, M. Jia, D. Jiang, K. Liu, R. Yu, Y. An, T. Pei and Y. Wang, J. Mater. Chem. A, 2026, 14, 3661–3668. 95 Z. Deng, T. P. Mishra, E. Mahayoni, Q. Ma, A. J. K. Tieu, O. Guillon, J.-N. Chotard, V. Seznec, A. K. Cheetham, C. Masquelier, G. S. Gautam and P. Canepa, Nat Commun, 2022, 13,
2013
-
[8]
96 A. S. Nogai, A. A. Nogai, S. Yu. Stefanovich, Zh. M. Solikhodzha and D. E. Uskenbaev, Phys. Solid State, 2020, 62, 1370–1379. 97 L. Pradhan and P. Padma Kumar, J. Phys. Chem. C, 2025, 129, 13756–13767. 98 S. Kim, M. Lee, C. Hong, Y. Yoon, H. An, D. Lee, W. Jeong, D. Yoo, Y. Kang, Y. Youn and S. Han, Sci Data, 2020, 7,
2020
-
[9]
99 J. M. Crowley, J. Tahir-Kheli and W. A. Goddard, J. Phys. Chem. Lett., 2016, 7, 1198–1203. 100 Z. Wang, T. P. Mishra, W. Xie, Z. Deng, G. S. Gautam, A. K. Cheetham and P. Canepa, DOI:10.48550/ARXIV.2308.04772
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.