Diagrammatic Multiplet-Sum Method (MSM) Density-Functional Theory(DFT): III. Inclusion of Relaxation and Application to LiH
Pith reviewed 2026-06-29 00:18 UTC · model grok-4.3
The pith
Incorporating NOCI relaxation into diagrammatic MSM DFT yields an accurate ground-state potential energy curve for LiH across its avoided crossing with charge transfer.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
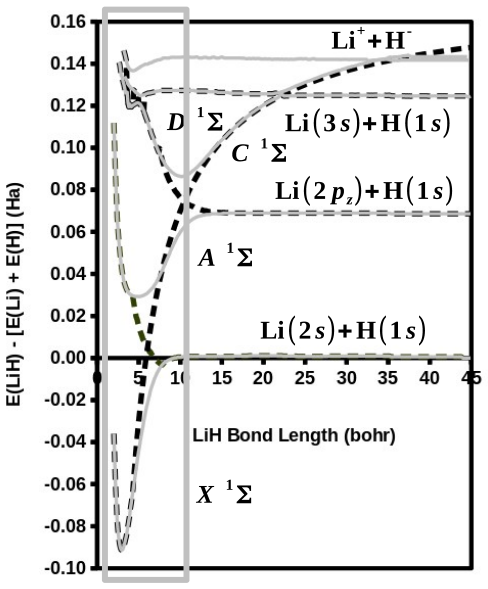
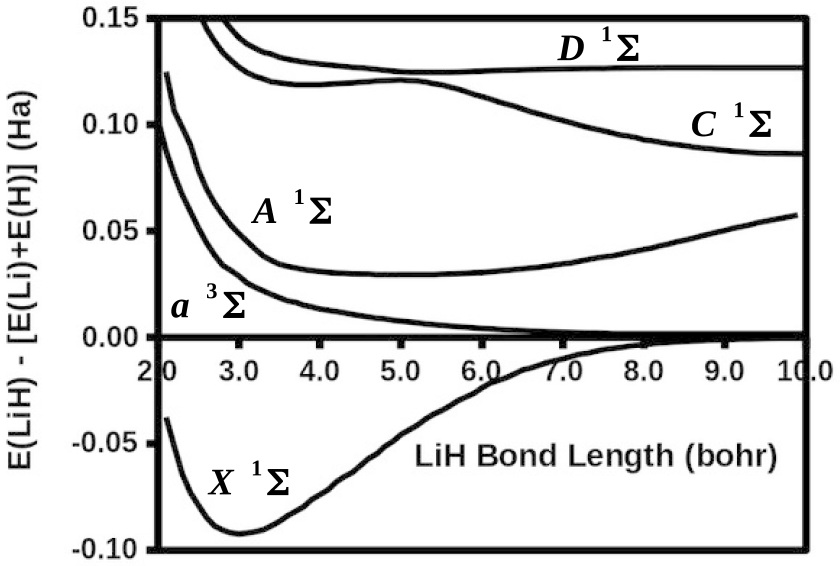
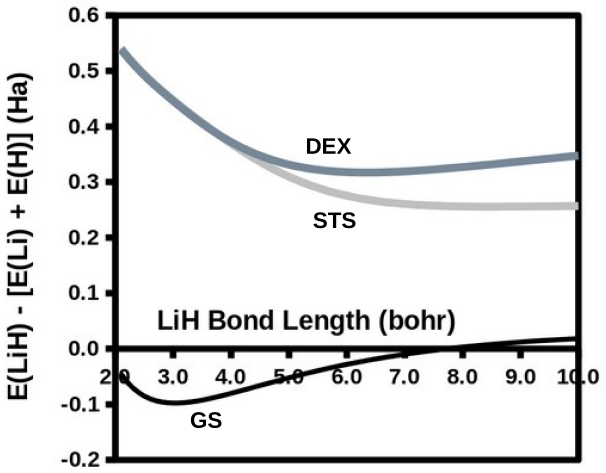
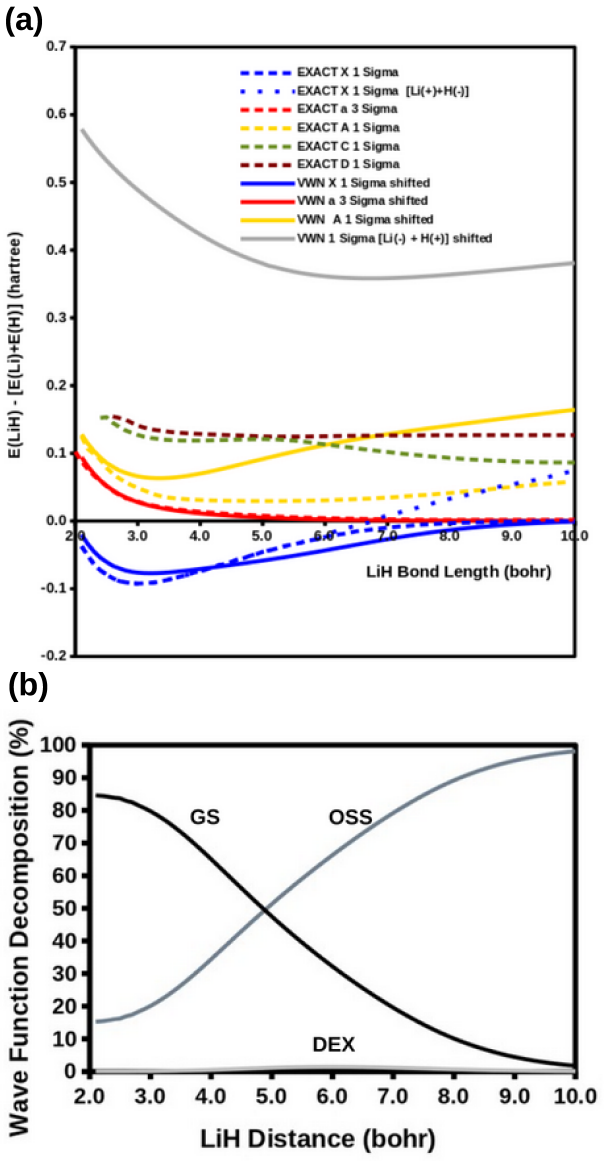
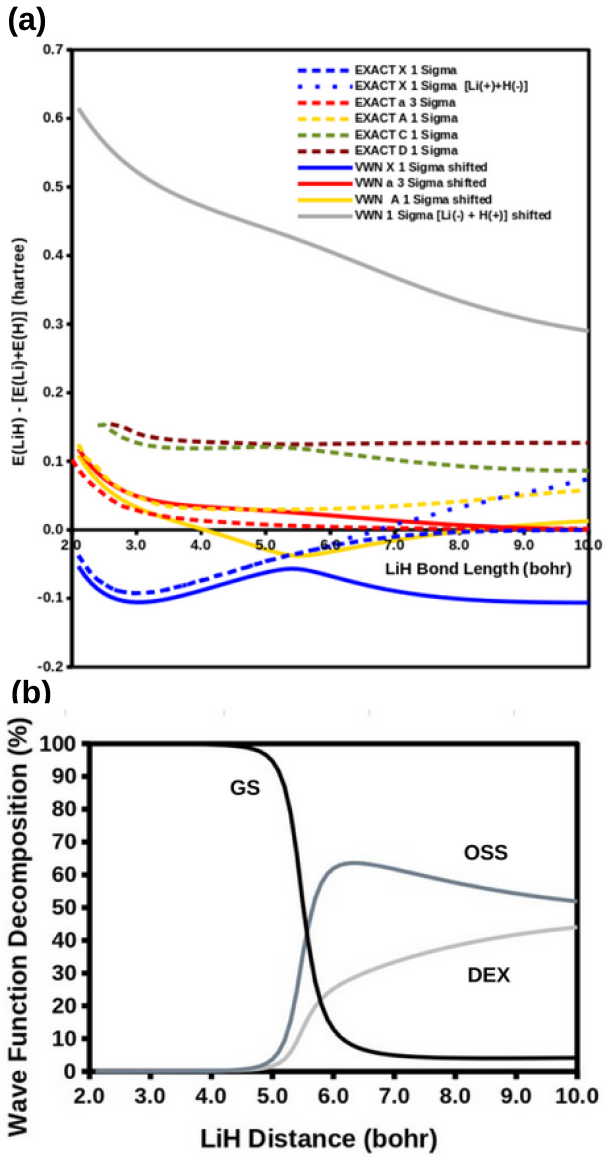
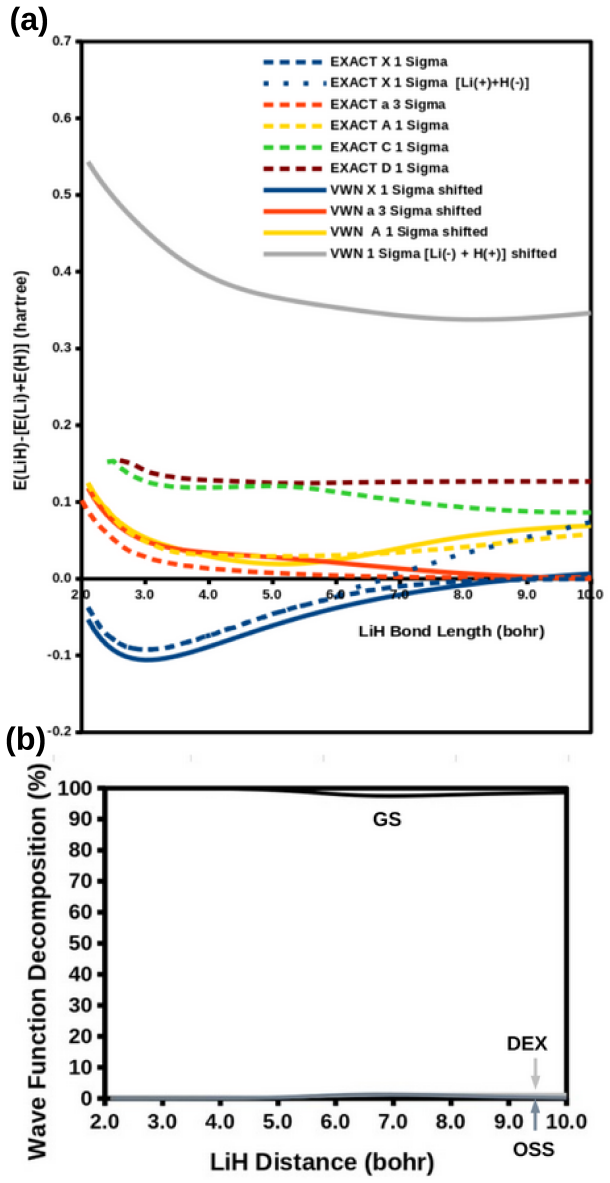
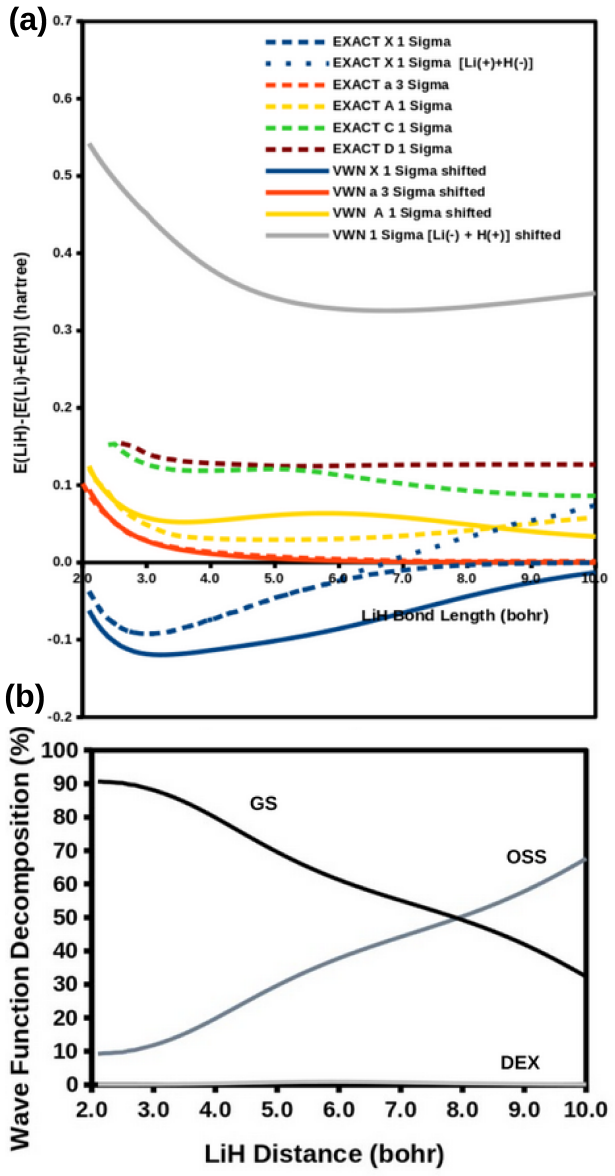
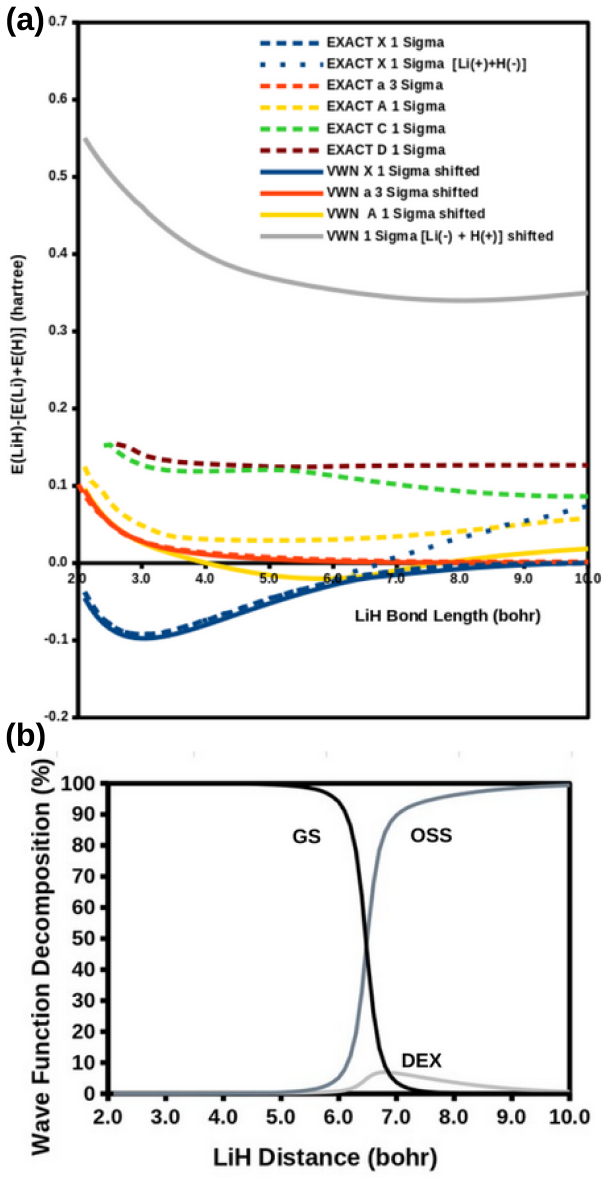
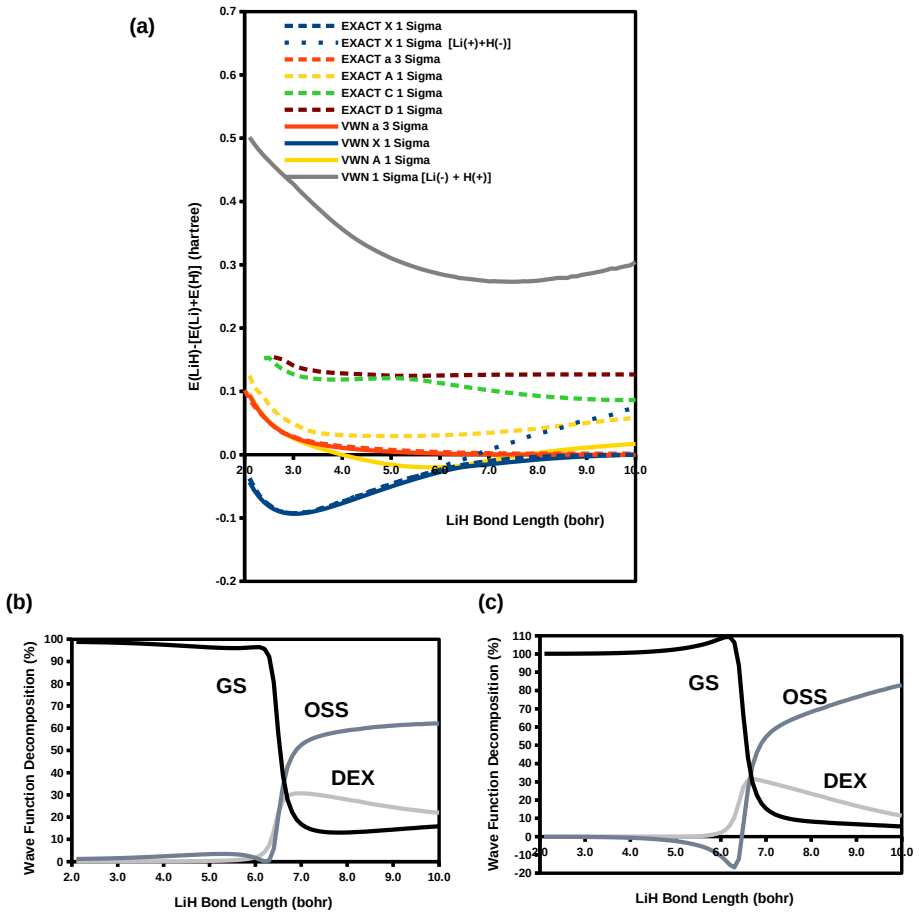
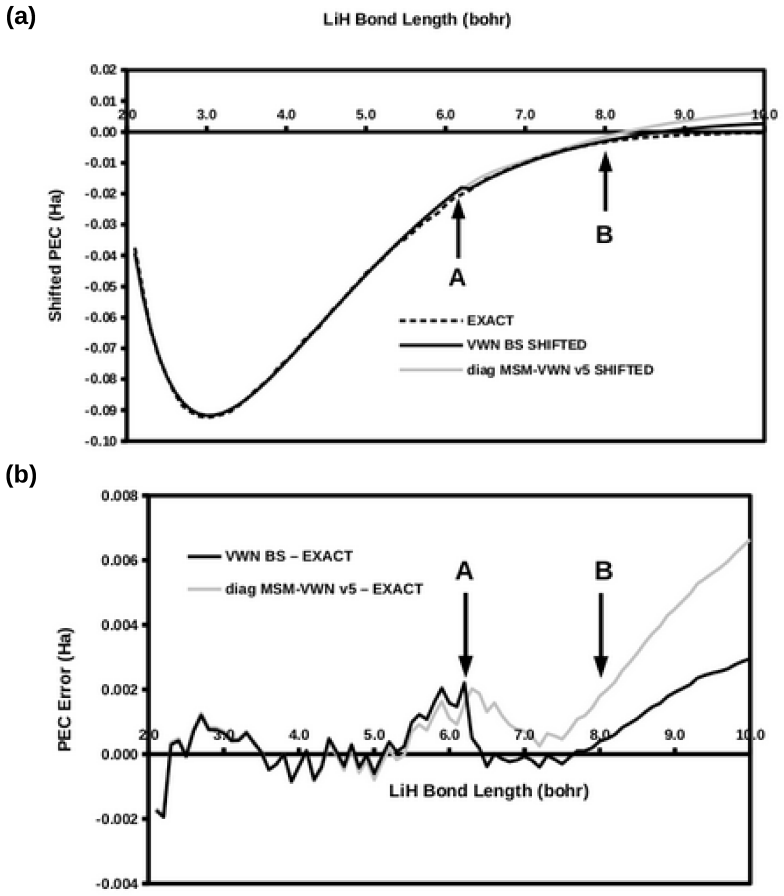
The modified diag MSM DFT using the two-orbital two-electron model together with diagrammatic multiplet sums and NOCI relaxation produces an accurate ground-state potential energy curve for LiH even at the ionic-to-open-shell-singlet avoided crossing characterized by significant charge transfer.
What carries the argument
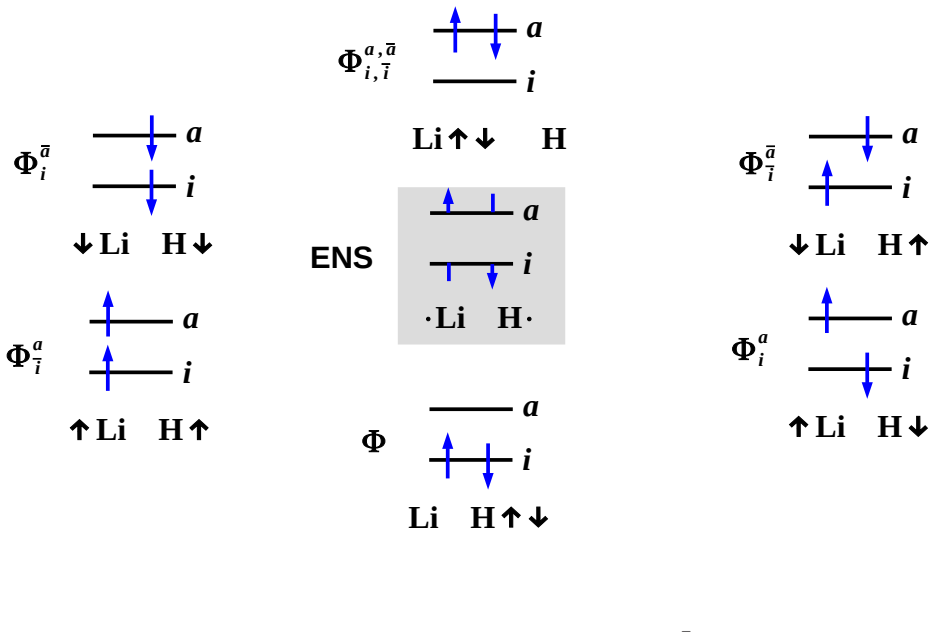
The two-orbital two-electron model (TOTEM) combined with diagrammatic multiplet sum method and nonorthogonal configuration interaction (NOCI) relaxation.
If this is right
- The approach accounts for dynamic, static, and nondynamic correlation within one density-functional framework.
- The method can be extended to at least other singly and multiply-bonded diatomic molecules.
- Nondynamic correlation is included without relying on symmetry arguments.
- Relaxation effects improve the description of charge-transfer regions in potential energy curves.
Where Pith is reading between the lines
- The same construction may apply to transition states or larger systems that exhibit avoided crossings driven by charge transfer.
- Direct numerical comparisons with multireference wavefunction methods could quantify the computational trade-offs.
- The framework might reduce reliance on empirical corrections for strong correlation in certain molecular classes.
Load-bearing premise
The two-orbital two-electron model together with diagrammatic MSM and NOCI relaxation is sufficient to capture the physics of the avoided crossing and charge transfer in LiH without additional fitted parameters or symmetry constraints.
What would settle it
High-level ab initio calculations or experimental data showing that the computed potential energy curve deviates markedly from reference values near the avoided crossing, either in energy or internuclear distance at the crossing point, would falsify the central claim.
Figures
read the original abstract
Ideal density-functional approximations (DFAs) should account for dynamic, static, and nondynamic correlation. While common DFAs struggle with the latter two, the Ziegler-Rauk-Baerends-Daul multiplet sum method (MSM) provides a pragmatic way to include static correlation. In this article, we use diagrammatic MSM density-functional theory (diag MSM DFT) using the two-orbital two-electron model (TOTEM) to extend MSM DFT to include nondynamic correlation without relying on symmetry arguments. Building on previous formulations [A. Ponra, C. Bakasa, A.J. Etindele,and M.E. Casida, J. Chem. Phys. 159, 244306 (2023); M.E. Casida, A. Ponra, C. Bakasa, and A.J.Etindele, J. Chem. Phys. 162, 144317 (2025)] that lacked relaxation effects, this article incorporates relaxation via nonorthogonal configuration interaction (NOCI). We demonstrate that this modified diag MSM DFT produces an accurate ground-state potential energy curve (PEC) for lithium hydride (LiH), even at the ionic-to-open-shell-singlet avoided crossing characterized by significant charge transfer. This encouraging result suggests that the model can be extended to (at least) other singly and multiply-bonded diatomic molecules, while providing insight into a novel way to include strong correlation in DFT.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript extends diagrammatic multiplet-sum method density-functional theory (diag MSM DFT) using the two-orbital two-electron model (TOTEM) to incorporate nondynamic correlation without symmetry arguments and adds relaxation via nonorthogonal configuration interaction (NOCI). It applies the resulting method to lithium hydride and claims that the modified approach produces an accurate ground-state potential energy curve even at the ionic-to-open-shell-singlet avoided crossing with significant charge transfer.
Significance. If the quantitative results support the claim, the work would supply a parameter-free route to static and nondynamic correlation in DFT for systems exhibiting avoided crossings and charge transfer, with possible extension to other singly and multiply bonded diatomics.
major comments (2)
- Abstract: the central claim that the modified diag MSM DFT 'produces an accurate ground-state potential energy curve (PEC) for lithium hydride (LiH)' is unsupported by any numerical values, error metrics, dissociation energies, equilibrium distances, or direct comparisons to experiment or reference methods, rendering the accuracy assertion at the avoided crossing unverifiable.
- Abstract: the reported accuracy for the LiH PEC rests on the two-orbital two-electron model (TOTEM) together with MSM and NOCI components defined in the authors' prior self-citations (J. Chem. Phys. 159, 244306 (2023) and 162, 144317 (2025)); the claim therefore reduces in part to quantities introduced in those earlier works without independent validation or sensitivity tests in the present manuscript.
minor comments (1)
- Title: 'Theory(DFT)' is missing a space before the parenthesis.
Simulated Author's Rebuttal
We thank the referee for their comments on our manuscript. We address each major comment below and indicate where revisions will be made to improve clarity and verifiability.
read point-by-point responses
-
Referee: Abstract: the central claim that the modified diag MSM DFT 'produces an accurate ground-state potential energy curve (PEC) for lithium hydride (LiH)' is unsupported by any numerical values, error metrics, dissociation energies, equilibrium distances, or direct comparisons to experiment or reference methods, rendering the accuracy assertion at the avoided crossing unverifiable.
Authors: We agree that the abstract would be strengthened by including quantitative support. The manuscript body contains figures and discussion comparing the computed PEC to reference data, but the abstract itself does not quote specific metrics. We will revise the abstract to report key values such as the dissociation energy, equilibrium distance, and the energy error at the avoided crossing relative to experiment and high-level reference calculations. revision: yes
-
Referee: Abstract: the reported accuracy for the LiH PEC rests on the two-orbital two-electron model (TOTEM) together with MSM and NOCI components defined in the authors' prior self-citations (J. Chem. Phys. 159, 244306 (2023) and 162, 144317 (2025)); the claim therefore reduces in part to quantities introduced in those earlier works without independent validation or sensitivity tests in the present manuscript.
Authors: The present manuscript introduces the combination of diag MSM DFT with NOCI relaxation and demonstrates its performance on LiH, including at the ionic-to-open-shell avoided crossing. While the underlying TOTEM, MSM, and NOCI formalisms are developed in the cited prior works, the current paper provides the first application and validation of the relaxed version to a charge-transfer avoided-crossing problem. We will add a brief paragraph in the introduction summarizing the key prior elements and their assumptions to make the manuscript more self-contained. Additional sensitivity tests could be included if specific parameters are identified as critical. revision: partial
Circularity Check
Self-citations define base method but central result is independent validation on LiH
full rationale
The paper extends prior diag MSM DFT (defined in two self-cited works) by adding NOCI relaxation and applies the result to compute the LiH ground-state PEC, claiming accuracy through the ionic-to-open-shell avoided crossing. This constitutes an empirical test against the known physical behavior of LiH rather than a derivation that reduces to its own inputs by construction. No equations or steps are shown where a prediction equals a fitted parameter, where an ansatz is smuggled via self-citation, or where a uniqueness theorem is invoked to force the outcome. The self-citations supply the starting formalism but do not bear the load of the new accuracy claim, which rests on the explicit inclusion of relaxation and the numerical comparison for this molecule. The derivation chain therefore remains self-contained against external benchmarks.
Axiom & Free-Parameter Ledger
axioms (1)
- domain assumption The two-orbital two-electron model (TOTEM) is adequate to describe the electronic structure changes at the ionic-to-open-shell-singlet avoided crossing in LiH.
Reference graph
Works this paper leans on
-
[1]
Hohenberg and W
P. Hohenberg and W. Kohn, Inhomogenous electron gas , Phys. Rev. 136, B864 (1964). 29
1964
-
[2]
Kohn and L
W. Kohn and L. J. Sham, Self-consistent equations including exchange and correla tion effects , Phys. Rev. 140, A1133 (1965)
1965
-
[3]
Seidl, A
A. Seidl, A. G¨ orling, P. Vogl, and J. A. Majewski, Generalized Kohn-Sham scheme and the band gap problem, Phys. Rev. B 53, 3764 (1996)
1996
-
[4]
R. J. Bartlett and J. F. Stanton, Applications of post-Hartree-Fock methods: A tutorial , in Reviews in Computational Chemistry, Vol. 5 , edited by K. B. Lipkowitz and D. B. Boyd, page 65, VCH Publishers, New York, 1994
1994
-
[5]
J. P. Perdew, R. G. Parr, M. Levy, and J. L. Balduz, Density-functional theory for fractional particle number: Derivative discontinuities of the energy , Phys. Rev. Lett. 49, 1691 (1982)
1982
-
[6]
L. J. Sham and M. Schl¨ uter, Density-functional theory of the energy gap , Phys. Rev. Lett. 51, 1888 (1983)
1983
-
[7]
Perdew, What do the Kohn-Sham orbital energies mean? How do atoms dis sociate?, in Density Functional Methods in Physics , edited by R
J. Perdew, What do the Kohn-Sham orbital energies mean? How do atoms dis sociate?, in Density Functional Methods in Physics , edited by R. M. Dreizler and J. da Providˆ encia, page 265, Plenum, New York, 1985
1985
-
[8]
O. V. Gritsenko and E. J. Baerends, Effect of molecular dissociation on the exchange-correlati on Kohn-Sham potential , Phys. Rev. A 54, 1957 (1996)
1957
-
[9]
D. G. Tempel, T. J. Mart ´ ınez, and N. T. Maitra, Revisiting molecular dissociation in density functional theory: A simple model , J. Chem. Theory Comput. 5, 770 (2009)
2009
-
[10]
M. E. Casida, F. Gutierrez, J. Guan, F. Gadea, D. R. Salahub, a nd J. Daudey, Charge-transfer correction for improved time-dependent local density appr oximation excited-state potential energy curves: Analysis within the two-level model with illustrat ion for H 2 and LiH , J. Chem. Phys. 113, 7062 (2000)
2000
-
[11]
Tapavicza, I
E. Tapavicza, I. Tavernelli, U. Rothlisberger, C. Filippi, and M. E. Casida, Mixed time-dependent density-functional theory/classical surface hopping stu dy of oxirane photochemistry , J. Chem. Phys. 129, 124108 (2008)
2008
-
[12]
A. D. Becke, Perspective: Fifty years of density-functional theory in c hemical physics , J. Chem. Phys. 140, 18A301 (2014)
2014
-
[13]
R. J. Bartlett, Ab initio DFT and its role in electronic structure theory , Mol. Phys. 108, 3299 (2010)
2010
-
[14]
Ziegler, A
T. Ziegler, A. Rauk, and E. J. Baerends, On the calculation of multiplet energies by the Hartree- Fock-Slater method , Theor. Chim. Acta 4, 877 (1977)
1977
-
[15]
Daul, Density functional theory applied to the excited states of c oordination compounds , Int
C. Daul, Density functional theory applied to the excited states of c oordination compounds , Int. J. Quantum Chem. 52, 867 (1994)
1994
-
[16]
Ponra, A
A. Ponra, A. J. Etindele, O. Motapon, and M. E. Casida, Practical treatment of singlet oxygen with density-functional theory and the multiplet-sum meth od, Theo. Chem. Acc. 140, 154 (2021)
2021
-
[17]
Ponra, C
A. Ponra, C. Bakasa, A. J. Etindele, and M. E. Casida, Diagrammatic multiplet-sum method (MSM) density-functional theory (DFT): Investigation of t he transferability of integrals in “simple” DFT-based approaches to multi-determinantal problems , J. Chem. Phys. 159, 244306 (2023). 30
2023
-
[18]
M. E. Casida, A. Ponra, C. Bakasa, and A. J. Etindele, Diagrammatic multiplet-sum method (MSM) density-functional theory (DFT): Completion of the two-orbital two-electron model (TOTEM) with an application to the avoided crossing in lithium hydri de (LiH) , J. Chem. Phys. 162, 144317 (2025)
2025
-
[19]
F. X. Gad´ ea and T. Leininger, Accurate ab initio calculations for LiH and its ions LiH + and LiH − , Theor. Chem. Acc. 116, 566 (2006)
2006
-
[20]
Rohatgi, WebPlotDigitizer, https://automeris.io/, Last accessed 18 February 2026
A. Rohatgi, WebPlotDigitizer, https://automeris.io/, Last accessed 18 February 2026
2026
-
[21]
Schaftenaar, MOLDEN: A pre- and post processing program of molecular electronic structure, https://www3.cmbi.umcn.nl/molden/, Last accessed 22 May 2021
B. Schaftenaar, MOLDEN: A pre- and post processing program of molecular electronic structure, https://www3.cmbi.umcn.nl/molden/, Last accessed 22 May 2021
2021
-
[22]
Lu and J
Y. Lu and J. Gao, Multistate density functional theory: Theory, methods, an d applications, WIREs Comput. Mol. Sci. 15, e70043 (2025)
2025
-
[23]
B. H. Chirgwin and C. A. Coulson, The electronic structure of conjugated systems. VI. , Proc. Roy. Soc. Lond. A 201, 196 (1950)
1950
-
[24]
L. H. Sarett, Research and invention , Proc. Natl. Acad. Sci. USA 80, 4572 (1983). 31
1983
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.