SVC-Probe: A Framework for Evaluating Perturbation Generalization in Spatial Foundation-Model Embeddings
Pith reviewed 2026-06-30 01:11 UTC · model grok-4.3
The pith
High accuracy distinguishing drug conditions in spatial embeddings does not imply reliable cross-drug prediction.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
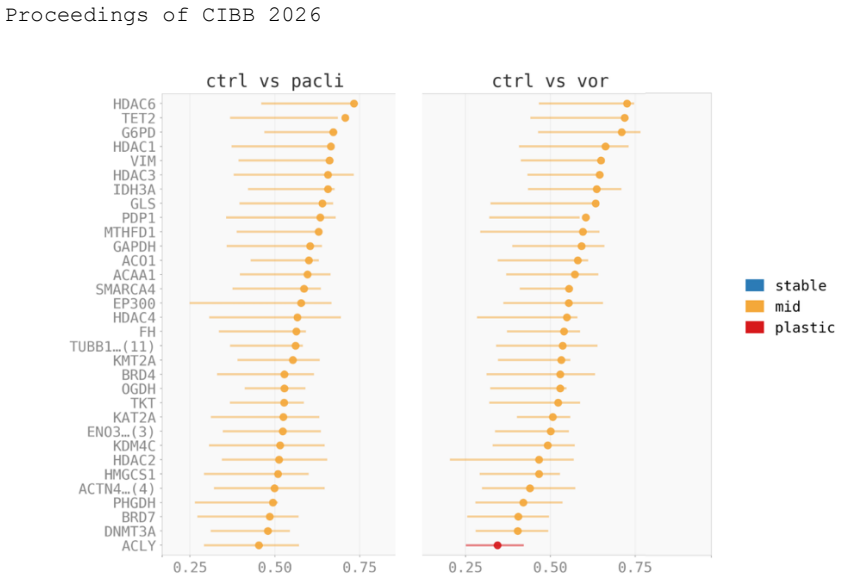
SVC-Probe demonstrates that 98.6% three-way condition accuracy does not correlate with reliable cross-drug prediction, with cosine similarity diminishing from 0.944 in-domain to 0.30 under leave-one-drug-out evaluation, constituting a two-drug stress test rather than a general benchmark. Null calibration indicates that raw residual-turnover coupling is largely influenced by generic embedding structure, whereas a drug-specific signal emerges under vorinostat and is consistent with chromatin-related reorganization. In contrast, the paclitaxel axis is not robustly reconstructed, likely due to sparse coverage of microtubule-associated proteins.
What carries the argument
SVC-Probe, a perturbation-aware framework that combines Subcellular Embedding Atlas Stability, Mondrian Neighborhood Graphs, and a Foundation Model Perturbation Probe to assess embedding stability, neighborhood rewiring, and centroid prediction under drug treatment.
If this is right
- Perturbation generalization serves as a stricter benchmark than baseline condition discrimination for spatial virtual-cell representations.
- Drug-specific signals can be isolated from generic embedding structure using null calibration.
- Sparse coverage of certain protein classes limits reconstruction of specific perturbation axes such as paclitaxel.
Where Pith is reading between the lines
- Applying the same probe to embeddings from other cell lines or perturbation classes could show whether the observed similarity drop is atlas-specific.
- Expanding antibody panels to include more microtubule-associated proteins would test if paclitaxel-axis reconstruction improves.
- Comparing multiple foundation models with SVC-Probe could quantify which architectures better capture transferable perturbation axes.
Load-bearing premise
That leave-one-drug-out evaluation on this particular atlas with its specific protein coverage provides a meaningful test of generalization to arbitrary perturbations.
What would settle it
Observing cosine similarity remaining above 0.7 in leave-one-drug-out tests on a new atlas with dense microtubule-associated protein labels would indicate the paclitaxel reconstruction failure is not due to sparsity.
Figures

read the original abstract
This work examines perturbation generalization in spatial foundation-model embeddings derived from fluorescence microscopy images. Although these models can discriminate drug conditions accurately, it remains unclear whether the learned representations reflect patterns consistent with expected perturbation axes that transfer across drugs. We introduce SVC-Probe, a perturbation-aware framework that combines Subcellular Embedding Atlas Stability, Mondrian Neighborhood Graphs, and a Foundation Model Perturbation Probe to assess embedding stability, neighborhood rewiring, and centroid prediction under drug treatment. Applied to the CM4AI MDA-MB-468 chemical-perturbation atlas comprising 462 antibody labels and SubCell 1536-dimensional embeddings, SVC-Probe demonstrates that 98.6% three-way condition accuracy does not correlate with reliable cross-drug prediction, with cosine similarity diminishing from 0.944 in-domain to 0.30 under leave-one-drug-out evaluation, constituting a two-drug stress test rather than a general benchmark. Null calibration indicates that raw residual-turnover coupling is largely influenced by generic embedding structure, whereas a drug-specific signal emerges under vorinostat and is consistent with chromatin-related reorganization. In contrast, the paclitaxel axis is not robustly reconstructed, likely due to sparse coverage of microtubule-associated proteins. Together, these results introduce and demonstrate a reusable diagnostic framework for stress-testing spatial virtual-cell representations and indicate that perturbation generalization may serve as a stricter and more informative benchmark than baseline condition discrimination.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript introduces SVC-Probe, a perturbation-aware framework that integrates Subcellular Embedding Atlas Stability, Mondrian Neighborhood Graphs, and a Foundation Model Perturbation Probe to evaluate embedding stability, neighborhood rewiring, and centroid prediction in spatial foundation-model embeddings from fluorescence microscopy. Applied to the CM4AI MDA-MB-468 chemical-perturbation atlas (462 antibody labels, SubCell 1536-dimensional embeddings), the work shows that 98.6% three-way condition accuracy does not imply reliable cross-drug prediction: cosine similarity falls from 0.944 (in-domain) to 0.30 (leave-one-drug-out). The evaluation is explicitly framed as a two-drug stress test; null calibration indicates mostly generic embedding structure, with a drug-specific chromatin signal only for vorinostat, while the paclitaxel axis fails due to sparse microtubule-protein coverage. The central claim is that perturbation generalization constitutes a stricter benchmark than baseline condition discrimination.
Significance. If the reported metrics and caveats hold, the paper supplies a reusable diagnostic framework for stress-testing spatial virtual-cell representations. The explicit positioning as a limited two-drug test, together with null calibration and protein-coverage caveats, provides a balanced assessment of current embedding limitations and highlights perturbation generalization as a more informative evaluation axis than simple classification accuracy.
minor comments (3)
- Abstract: the quantitative claims (98.6% accuracy, cosine values 0.944/0.30) would benefit from a parenthetical reference to the exact data split and embedding dimensionality used, even at abstract length.
- Methods/Results: clarify whether the Mondrian Neighborhood Graphs component includes any tunable parameters (e.g., neighborhood size) and, if so, how they were chosen or shown to be robust.
- Results: the null-calibration section would be strengthened by an explicit table or figure panel contrasting generic vs. drug-specific signals across all tested perturbations rather than narrative description alone.
Simulated Author's Rebuttal
We thank the referee for their positive and accurate summary of the manuscript, which correctly captures the scope of SVC-Probe, the reported metrics, the two-drug stress-test framing, and the protein-coverage caveats. We appreciate the recommendation for minor revision. No specific major comments were provided in the report.
Circularity Check
No significant circularity identified
full rationale
The paper introduces SVC-Probe as an evaluation framework applied to an external atlas (CM4AI MDA-MB-468) using explicit data splits such as leave-one-drug-out. No equations, fitted parameters, self-citations, or ansatzes are presented that reduce the reported metrics (condition accuracy, cosine similarity) to inputs by construction. The central results derive from standard cross-validation on independent data rather than self-referential definitions or renamings.
Axiom & Free-Parameter Ledger
Reference graph
Works this paper leans on
-
[1]
A subcellular map of the human proteome,
Thul et al., “A subcellular map of the human proteome,” Science, vol. 356, p. eaal3321, 2017
2017
-
[2]
SubCell: Proteome-aware vision foundation models for microscopy capture single-cell biology,
Gupta et al., “SubCell: Proteome-aware vision foundation models for microscopy capture single-cell biology,” bioRxiv 2024.12.06.627299, 2024. Proceedings of CIBB 2026 7
2024
-
[3]
A foundation model for spatial proteomics,
Shaban et al., “A foundation model for spatial proteomics,” arXiv:2506.03373, 2025
-
[4]
scGen predicts single-cell perturbation responses,
Lotfollahi et al., “scGen predicts single-cell perturbation responses,” Nat. Methods, vol. 16, pp. 715–721, 2019
2019
-
[5]
Learning single-cell perturbation responses using neural optimal transport,
Bunne et al., “Learning single-cell perturbation responses using neural optimal transport,” Nat. Methods, vol. 20, pp. 1759–1768, 2023
2023
-
[6]
Predicting transcriptional outcomes of novel multigene perturbations with GEARS,
Roohani et al., “Predicting transcriptional outcomes of novel multigene perturbations with GEARS,” Nat. Biotechnol., vol. 42, pp. 927–935, 2024
2024
-
[7]
Coordinated cellular neighborhoods orchestrate antitumoral immunity at the colorectal cancer invasive front,
Schürch et al., “Coordinated cellular neighborhoods orchestrate antitumoral immunity at the colorectal cancer invasive front,” Cell, vol. 182, pp. 1341–1359, 2020
2020
-
[8]
The proliferation rate paradox in antimitotic chemotherapy,
Mitchison, “The proliferation rate paradox in antimitotic chemotherapy,” Mol. Biol. Cell, vol. 23, pp. 1 –6, 2012
2012
-
[9]
New and emerging HDAC inhibitors for cancer treatment,
West et al., “New and emerging HDAC inhibitors for cancer treatment,” J. Clin. Invest., vol. 124, pp. 30 –39, 2014
2014
-
[10]
Cell Maps for Artificial Intelligence: AI -ready maps of human cell architecture from disease - relevant cell lines,
Clark et al., “Cell Maps for Artificial Intelligence: AI -ready maps of human cell architecture from disease - relevant cell lines,” bioRxiv 2024.05.21.589311, 2024
2024
-
[11]
Densely connected convolutional networks,
Huang et al., “Densely connected convolutional networks,” in Proc. CVPR, pp. 4700–4708, 2017
2017
-
[12]
Generative machine learning unlocks the first proteome -wide image of human cells,
Sun et al., “Generative machine learning unlocks the first proteome -wide image of human cells,” bioRxiv 2026.03.31.715748, 2026
2026
-
[13]
scPortrait integrates single -cell images into multimodal modeling,
Mädler et al., “scPortrait integrates single -cell images into multimodal modeling,” bioRxiv 2025.09.22.677590, 2025
2025
-
[14]
Deep learning-based gene perturbation effect prediction does not yet outperform simple linear methods,
Ahlmann-Eltze et al., “Deep learning-based gene perturbation effect prediction does not yet outperform simple linear methods,” Nat. Methods, vol. 22, pp. 1657–1661, 2025
2025
-
[15]
Cellpose 2.0: how to train your own model,
Pachitariu et al., “Cellpose 2.0: how to train your own model,” Nat. Methods, vol. 19, pp. 1634–1641, 2022
2022
-
[16]
From Louvain to Leiden: guaranteeing well -connected communities,
Traag et al., “From Louvain to Leiden: guaranteeing well -connected communities,” Sci. Reports, vol. 9, p. 5233, 2019
2019
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.