A wrong ground-state structure of HfO₂ predicted by machine-learning interatomic potentials based on the PBE functional
Pith reviewed 2026-06-27 06:33 UTC · model grok-4.3
The pith
PBE-based machine-learning potentials predict an incorrect ground-state structure for HfO2.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
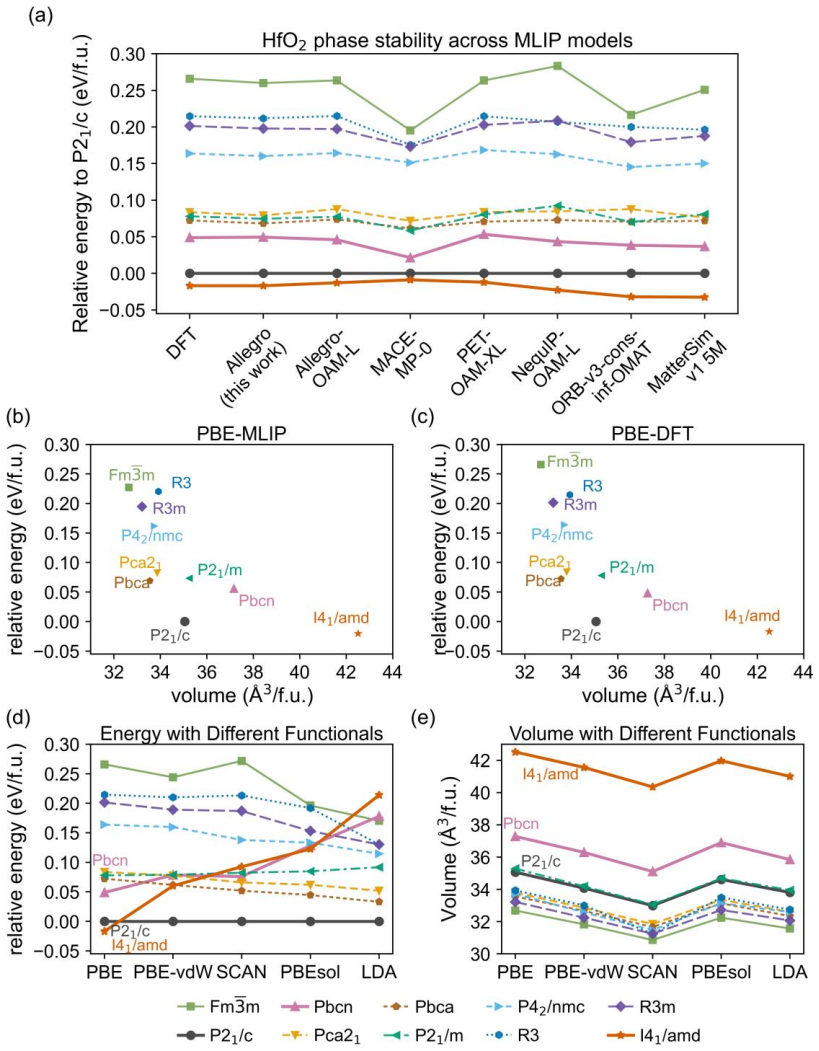
Using a PBE-trained MLIP for HfO2 yields a previously unreported I41/amd structure that lies lower in energy than the monoclinic P21/c phase known from experiment to be the ground state. Direct comparisons across DFT functionals establish that PBE itself favors these low-density sixfold-coordinated structures, and the same ordering error propagates into MLIP predictions as well as into the calculated ferroelectric switching paths that involve substantial lattice changes. The ordering is corrected when the underlying data are generated with PBEsol or LDA instead.
What carries the argument
PBE-trained machine-learning interatomic potential that reproduces the functional's erroneous energy ordering among HfO2 polymorphs containing sixfold Hf-O units.
If this is right
- The same incorrect ground-state prediction is made by other PBE-based foundation models such as NequIP-OAM-L and MatterSim-v1-5M.
- Energy landscapes and barrier heights along ferroelectric polarization switching paths are distorted whenever large lattice relaxations occur.
- The stability ordering error is largely removed when the training data come from PBEsol or local-density-approximation functionals.
- Simulations of crystal structures and phase transitions in HfO2 that rely on PBE-based MLIPs require extra validation against experiment.
Where Pith is reading between the lines
- The same PBE bias may affect MLIP predictions for other oxides whose low-density phases contain sixfold-coordinated metal-oxygen units.
- Training MLIPs on datasets generated with multiple functionals could reduce the risk of inheriting single-functional errors.
- For HfO2 phase-transition studies, potentials fitted to PBEsol data would avoid the reported stability reversal.
- Even high-accuracy MLIPs still inherit the fundamental limitations of the exchange-correlation functional used to generate their training data.
Load-bearing premise
The machine-learning interatomic potential accurately reproduces the PBE energy surface without adding fitting artifacts of its own.
What would settle it
A direct PBE density-functional calculation that places the P21/c structure lower in energy than I41/amd, or an MLIP whose energies deviate markedly from the underlying PBE surface on these two structures.
Figures
read the original abstract
Machine-learning interatomic potentials (MLIPs) have become powerful tools for material simulations. Many MLIPs are trained based on density functional theory (DFT) datasets generated with the Perdew-Burke-Ernzerhof (PBE) exchange-correlation functional. Using a PBE-based MLIP for HfO2, we identify a previously unreported low-energy I41/amd structure, which is predicted to be more stable than the well-known ground-state structure, the monoclinic P21/c structure. Since experiments show clearly that HfO2 takes the P21/c structure as the ground state, this is obviously a wrong prediction. Unfortunately, the same prediction is also made by widely used PBE-based foundation models such as NequIP-OAM-L and MatterSim-v1-5M. Comparisons among different DFT functionals show that this error originates from the PBE functional, which overstabilizes low-density structures containing sixfold Hf-O octahedral units, such as the I41/amd and Pbcn phases. The error also affects the calculated energy landscapes and barrier heights along ferroelectric HfO2 polarization switching paths when there are large lattice relaxations. Fortunately, the error can be largely suppressed by other functionals such as PBEsol and local density approximation. Our study serves as a warning about the impact of errors in exchange-correlation functional approximations on the reliability of MLIP simulations of crystal structures and phase transitions.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript claims that PBE-based MLIPs for HfO₂ predict an incorrect ground state (I41/amd lower in energy than experimental P21/c), with the same error appearing in foundation models such as NequIP-OAM-L and MatterSim-v1-5M. The authors attribute the error to the PBE functional overstabilizing low-density sixfold Hf-O octahedral structures (I41/amd, Pbcn), supported by comparisons showing that PBEsol and LDA reverse the ordering. The error is also shown to affect ferroelectric switching energy landscapes with large lattice relaxations.
Significance. If the result holds, the work is significant because it isolates a functional-level origin for a concrete failure in structure prediction for a technologically relevant material, rather than blaming the MLIP architecture. The direct multi-functional comparisons and checks on widely used foundation models are strengths that make the finding actionable for the community. The paper supplies a falsifiable test (energy ordering under different functionals) and reproducible guidance on mitigation.
major comments (2)
- [Results section describing MLIP structure search and energy ordering] The central claim that the I41/amd–P21/c ordering error originates from PBE (rather than an MLIP fitting artifact) is load-bearing and requires explicit verification. Direct PBE DFT total-energy differences between the relaxed I41/amd and P21/c structures must be reported and shown to match the MLIP ordering; the current DFT comparisons among functionals do not automatically confirm that the specific MLIP reproduces the PBE surface on these two phases.
- [Section on ferroelectric polarization switching] § on ferroelectric switching paths: the statement that the error 'affects the calculated energy landscapes and barrier heights' needs quantitative support. The magnitude of the change in barrier height when switching from PBE to PBEsol (or LDA) should be given for at least one path that involves large lattice relaxation, so readers can judge whether the effect is practically significant.
minor comments (2)
- [Results] Table or figure reporting the relaxed lattice parameters and volumes of I41/amd, Pbcn, and P21/c under each functional would improve clarity and allow direct comparison with experiment.
- [Methods] The training-set composition for the custom PBE MLIP (number of structures, sampling of sixfold vs. sevenfold coordination) should be stated explicitly to address possible under-sampling of the I41/amd phase.
Simulated Author's Rebuttal
We thank the referee for the constructive comments and for recognizing the significance of isolating a functional-level origin for the observed failure in structure prediction. We address each major comment below.
read point-by-point responses
-
Referee: [Results section describing MLIP structure search and energy ordering] The central claim that the I41/amd–P21/c ordering error originates from PBE (rather than an MLIP fitting artifact) is load-bearing and requires explicit verification. Direct PBE DFT total-energy differences between the relaxed I41/amd and P21/c structures must be reported and shown to match the MLIP ordering; the current DFT comparisons among functionals do not automatically confirm that the specific MLIP reproduces the PBE surface on these two phases.
Authors: We agree that explicit verification is required to confirm the MLIP reproduces the PBE surface on the specific phases. In the revised manuscript we will add direct PBE DFT total-energy differences (computed on the MLIP-relaxed I41/amd and P21/c structures) and demonstrate that the ordering matches the MLIP prediction, thereby confirming the functional origin of the error. revision: yes
-
Referee: [Section on ferroelectric polarization switching] § on ferroelectric switching paths: the statement that the error 'affects the calculated energy landscapes and barrier heights' needs quantitative support. The magnitude of the change in barrier height when switching from PBE to PBEsol (or LDA) should be given for at least one path that involves large lattice relaxation, so readers can judge whether the effect is practically significant.
Authors: We agree that quantitative values are needed for readers to assess practical significance. In the revised manuscript we will report the magnitude of the barrier-height change (PBE versus PBEsol or LDA) for at least one ferroelectric switching path that involves large lattice relaxation. revision: yes
Circularity Check
No significant circularity; claim rests on direct DFT functional comparisons
full rationale
The paper's central claim—that PBE overstabilizes sixfold Hf-O structures—is supported by explicit comparisons of multiple DFT functionals (PBE, PBEsol, LDA) on the I41/amd, Pbcn, and P21/c structures, benchmarked against experiment. No load-bearing step reduces to a fitted parameter renamed as prediction, self-citation chain, or self-definitional equation. The MLIP serves only as a discovery tool; the functional error is verified independently. This matches the default expectation of self-contained, non-circular analysis.
Axiom & Free-Parameter Ledger
axioms (2)
- domain assumption The monoclinic P21/c structure is the experimental ground state of HfO2
- domain assumption An MLIP trained on PBE data reproduces the PBE energy landscape sufficiently accurately that any wrong ordering can be attributed to PBE rather than to the fitting procedure
Reference graph
Works this paper leans on
-
[1]
MatterSim: A Deep Learning Atomistic Model Across Elements, Temperatures and Pressures
1 Schütt, K. T., Sauceda, H. E., Kindermans, P.-J., Tkatchenko, A. & Müller, K.-R. SchNet: a deep learning architecture for molecules and materials. J. Chem. Phys. 148, 241722 (2018). 2 Schütt, K. et al. SchNet: a continuous-filter convolutional neural network for modeling quantum interactions. Adv. Neural Inf. Process. Syst. 30, 992–1002 (2017). 3 Zhang,...
work page internal anchor Pith review Pith/arXiv arXiv doi:10.5281/zenodo.17087883 2018
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.