Cepstral Analysis to accelerate Green-Kubo thermal conductivity calculations of Metal-Organic Frameworks
Pith reviewed 2026-06-27 05:55 UTC · model grok-4.3
The pith
Cepstral analysis with Green-Kubo simulations gives stable thermal conductivity values for metal-organic frameworks after 1-2 ns of sampling.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
Cepstral analysis in combination with Green-Kubo simulations provides a robust route to massively mitigate statistical noise and ambiguous user-defined parameters while simultaneously reducing the required sampling times, yielding stable results across a wide range of correlation lengths and achieving convergence within about 1-2 ns of total sampling time for MOF-5, HKUST-1, and ZIF-8.
What carries the argument
Cepstral analysis of the heat-current autocorrelation function from equilibrium molecular dynamics, which extracts the thermal conductivity from the zero-frequency component after transformation of the noisy time series.
If this is right
- Results remain stable across a wide range of correlation lengths.
- Convergence occurs within 1-2 ns of total sampling time.
- The approach works with machine-learned moment tensor potentials trained on DFT data.
- It forms the basis for an automation-ready framework for near ab initio thermal transport predictions in MOFs and other low-conductivity materials.
Where Pith is reading between the lines
- The same cepstral processing could be tested on other porous or disordered materials where Green-Kubo noise is severe.
- Shorter sampling times may enable screening of much larger MOF databases for applications in which heat transport limits performance.
- The method might be combined with enhanced sampling techniques to study even larger or more complex frameworks.
Load-bearing premise
Cepstral analysis recovers the true thermal conductivity from the noisy autocorrelation data without introducing systematic bias.
What would settle it
Independent non-equilibrium molecular dynamics runs or experimental measurements on MOF-5, HKUST-1, or ZIF-8 that differ substantially from the cepstral Green-Kubo values.
Figures

read the original abstract
Metal-organic frameworks (MOFs) are promising porous materials for applications such as gas storage and separation, where heat transport can critically affect device performance. However, reliable computational prediction of their thermal conductivities remains challenging. In particular, equilibrium molecular-dynamics-based Green-Kubo (GK) simulations, as the most widely used approach, are severely affected by statistical noise. Moreover, they rely on multiple ambiguous, user-defined parameters, which hinder transferability and automation. Here, we demonstrate for metal-organic frameworks that cepstral analysis in combination with GK simulations provides a robust route to massively mitigate these problems, while simultaneously reducing the required sampling times. This is shown for three prototypical frameworks, MOF-5, HKUST-1, and ZIF-8, employing machine-learned moment tensor potentials trained on DFT reference data. In contrast to conventional, direct GK analysis, which shows erratic convergence and strong sensitivity to ad hoc choices of parameters, the cepstral approach yields stable results across a wide range of correlation lengths and achieves convergence within about 1-2 ns of total sampling time. This establishes cepstral analysis base Green-Kubo simulations combined with machine-learned potentials as an efficient, reproducible and automation-ready framework for near ab initio accuracy prediction of thermal transport in MOFs and other complex low-thermal-conductivity materials.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript claims that cepstral analysis applied to Green-Kubo (GK) heat-current autocorrelation functions from equilibrium MD simulations (using machine-learned moment tensor potentials trained on DFT data) yields stable, reproducible thermal conductivity values for MOF-5, HKUST-1, and ZIF-8. It asserts that this approach mitigates statistical noise and user-defined parameter sensitivity of direct GK integration while achieving convergence within 1-2 ns of total sampling time.
Significance. If the cepstral estimates are unbiased and match the true long-time GK limit, the method would offer a practical advance for automated, lower-cost thermal transport calculations in low-conductivity porous materials, supporting applications in gas storage and separation.
major comments (1)
- [Abstract and Results] The central claim that cepstral analysis extracts the physically correct thermal conductivity (without systematic bias from damping of long-time tails) rests only on demonstrated stability across correlation lengths and faster apparent convergence; no comparisons to independent methods (NEMD, ultra-long direct GK, or analytic limits) are provided to confirm absence of bias (abstract and results sections).
minor comments (1)
- [Methods] The description of the cepstral filter implementation (e.g., exact form of the filter, handling of the zero-frequency component) should be expanded with explicit equations or pseudocode for full reproducibility.
Simulated Author's Rebuttal
We thank the referee for their constructive review. We respond to the single major comment below.
read point-by-point responses
-
Referee: [Abstract and Results] The central claim that cepstral analysis extracts the physically correct thermal conductivity (without systematic bias from damping of long-time tails) rests only on demonstrated stability across correlation lengths and faster apparent convergence; no comparisons to independent methods (NEMD, ultra-long direct GK, or analytic limits) are provided to confirm absence of bias (abstract and results sections).
Authors: The referee correctly observes that the manuscript does not contain direct benchmarks against NEMD, ultra-long direct GK, or analytic limits. However, the central claim does not rest solely on numerical stability. Cepstral analysis estimates the thermal conductivity from the zero-frequency intercept of the cepstrum of the heat-current time series. This procedure recovers the full integral of the autocorrelation function from its frequency-domain representation without imposing an explicit time cutoff or exponential damping, thereby avoiding the truncation bias that affects direct integration. The observed invariance of the result over a wide range of maximum correlation lengths is a direct consequence of this property: any systematic damping of long-time tails would produce a detectable dependence on the correlation length, which is absent. The mathematical foundation and prior validation of the cepstral approach for Green-Kubo calculations are referenced in the manuscript. We therefore maintain that the evidence presented is sufficient to support the stated conclusions. revision: no
Circularity Check
No circularity: empirical demonstration of cepstral filtering on GK autocorrelations
full rationale
The paper applies an established cepstral technique (from signal processing) to the heat-current autocorrelation function obtained from equilibrium MD. The central results are empirical: stability of the integrated conductivity across correlation lengths and faster apparent convergence (1-2 ns) versus direct GK. These are direct numerical comparisons on three MOFs using ML potentials; no derivation reduces the reported conductivity value to a fitted parameter or to a self-citation by construction. No self-definitional steps, no 'prediction' that is the input renamed, and no load-bearing uniqueness theorem imported from the authors' prior work. The method is externally falsifiable against longer direct GK runs or NEMD, satisfying the independence criteria.
Axiom & Free-Parameter Ledger
Reference graph
Works this paper leans on
-
[1]
non -equilibrium molecular dynamics
Introduction Metal–organic frameworks (MOFs) have garnered intense research interest due to their high porosity and structural tunability. The latter arises from their hybrid organic -inorganic architecture, which combines organic linker molecules with metal ion nodes or clusters, leading to an effectively unlimited number of possible structures. 1 Numero...
-
[2]
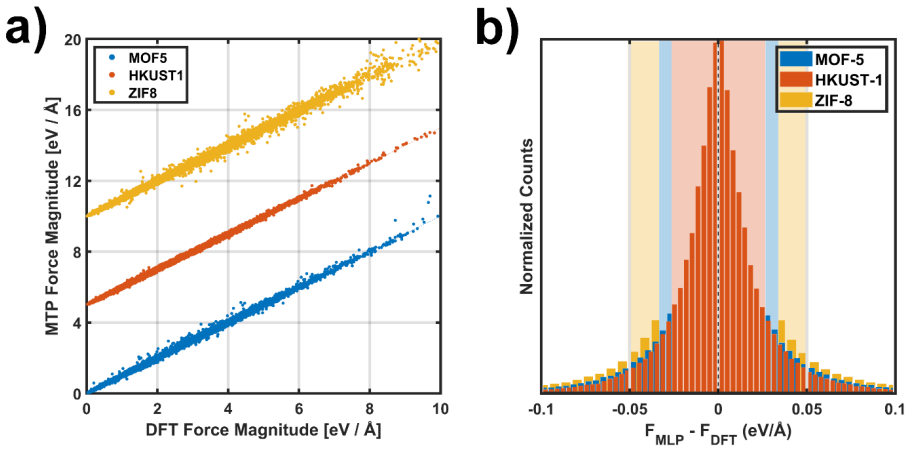
Computational Details 2.1. DFT calculations All DFT calculations (used for generating reference data for training the machine -learned potentials) were performed using the VASP code.57–59 Unless otherwise specified, a plane-wave energy cut-off of 900 eV and a Γ -point-only k-point sampling were employed. The latter is justified by the flat electronic band...
-
[3]
i”, vi is the velocity of atom “i
Fundamental Methodological Aspects Based on the fluctuation dissipation theorem,73 the GK equations connect spontaneo us fluctuations of the heat flux in thermal equilibrium to transport under non -equilibrium conditions. In this chapter, we first summarize the main concepts of the GK theory and then recapitulate how the exact equations are approximated i...
-
[4]
standard
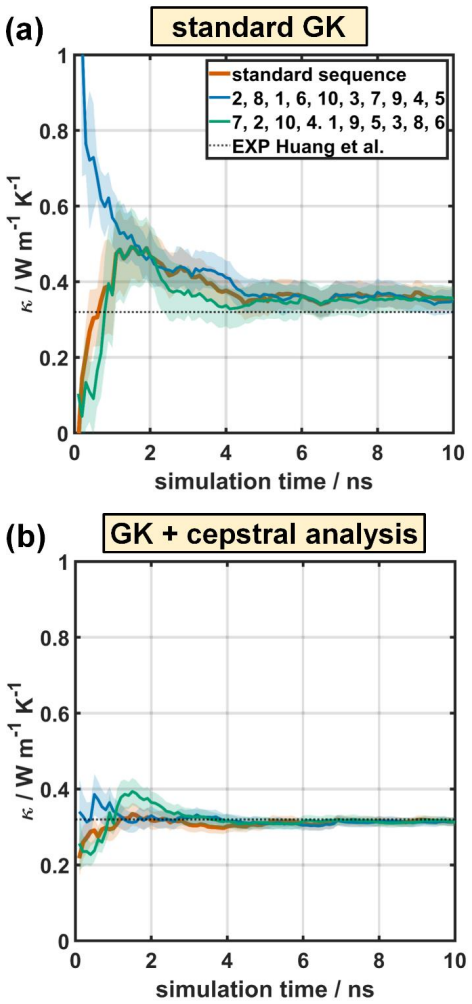
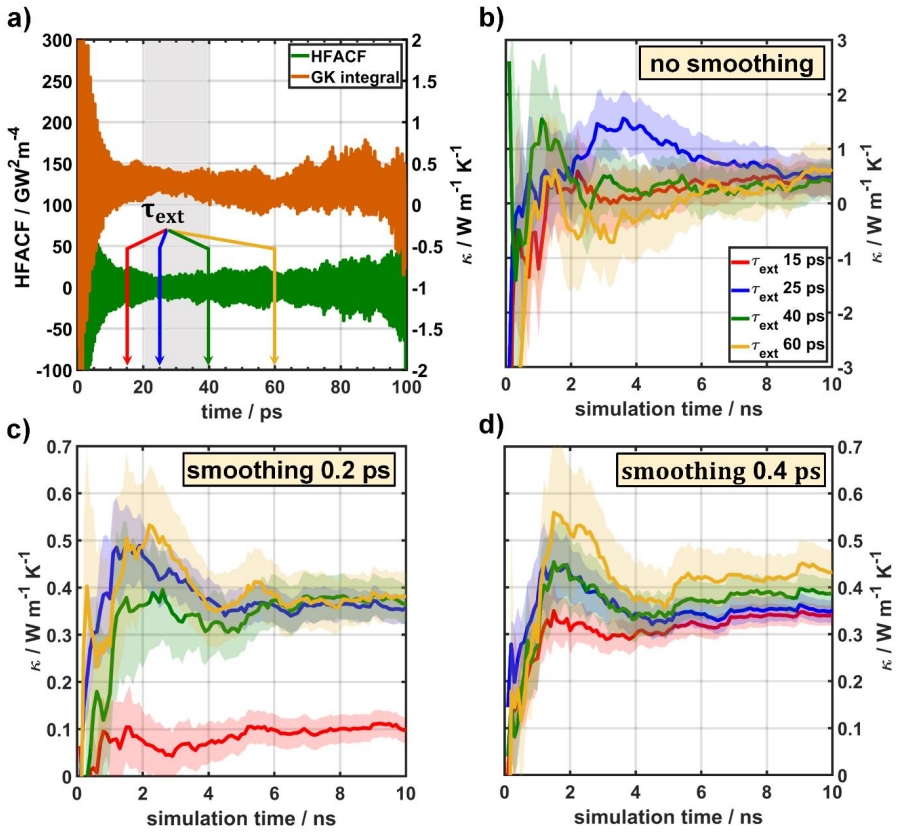
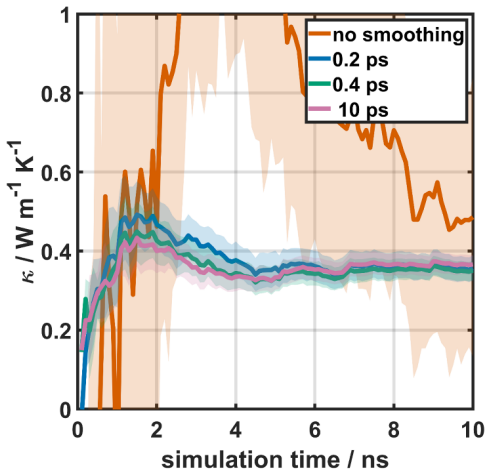
Results and Discussion The previous sections describe the working principle for standard GK simulations and for simulations augmented by cepstral analysis; in the following, the impact of various ambiguous process parameters on the results shall be discussed for the MOF-5 case considering trajectories with a total simulation time of 10 ns. Finally, also t...
-
[5]
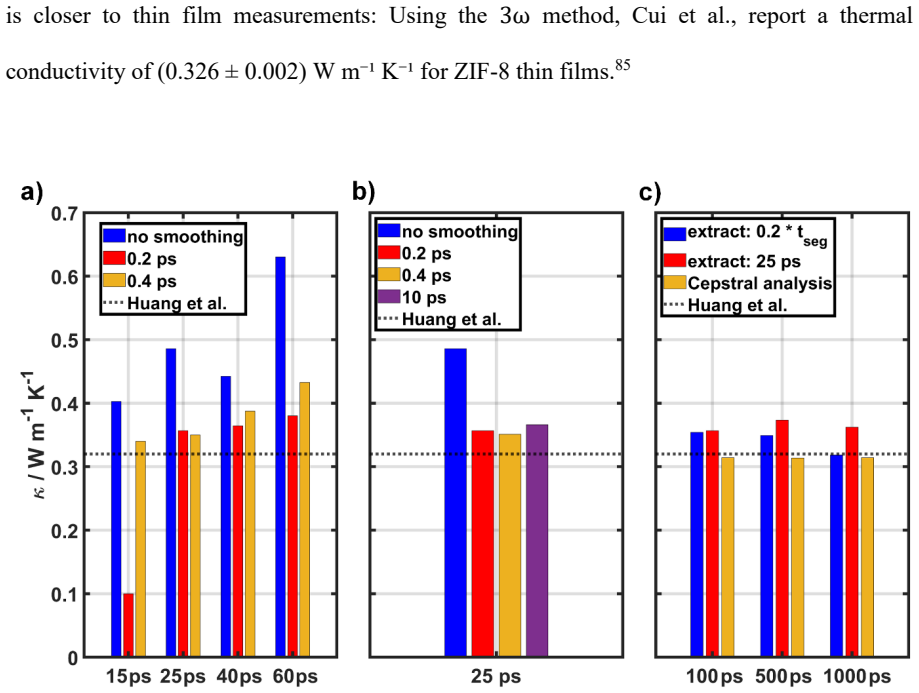
Conclusion In this work, we assessed the applicability of cepstral analysis for reducing the impact of noise when evaluating GK integrals for determining thermal conductivities in MOFs. We explicitly demonstrated how user defined ad -hoc parameters introduced along the simulation workflow of “standard GK” simulations can massively influence the obtained r...
-
[6]
Moghadam, P. Z. et al. Development of a Cambridge Structural Database Subset: A Collection of Metal–Organic Frameworks for Past, Present, and Future. Chem. Mater. 29, 2618–2625 (2017)
2017
-
[7]
Li, T. et al. Scalable and efficient solar-driven atmospheric water harvesting enabled by bidirectionally aligned and hierarchically structured nanocomposites. Nat. Water 1, 971–981 (2023)
2023
-
[8]
Shock wave energy absorption via structural phase transition and bond breakage in metal–organic frameworks
Banlusan, K. Shock wave energy absorption via structural phase transition and bond breakage in metal–organic frameworks. J. Chem. Phys. 163, 014703 (2025)
2025
-
[9]
Sun, Y. et al. High-rate nanofluidic energy absorption in porous zeolitic frameworks. Nat. Mater. 20, 1015–1023 (2021)
2021
-
[10]
Shupletsov, L. et al. Linker Conformation Controls Oxidation Potentials and Electrochromism in Highly Stable Zr-Based Metal–Organic Frameworks. J. Am. Chem. Soc. 146, 25477–25489 (2024)
2024
-
[11]
Xu, X. et al. Iontronics Using V2CTx MXene-Derived Metal–Organic Framework Solid Electrolytes. ACS Nano 14, 9840–9847 (2020)
2020
-
[12]
& Kaskel, S
Senkovska, I., Bon, V., Mosberger, A., Wang, Y. & Kaskel, S. Adsorption and Separation by Flexible MOFs. Adv. Mater. 37, 2414724 (2025)
2025
-
[13]
Lin, J.-B. et al. A scalable metal-organic framework as a durable physisorbent for carbon dioxide capture. Science 374, 1464–1469 (2021)
2021
-
[14]
Wieme, J. et al. Thermal Engineering of Metal–Organic Frameworks for Adsorption Applications: A Molecular Simulation Perspective. ACS Appl. Mater. Interfaces 11, 38697–38707 (2019)
2019
-
[15]
& Dailly, A
Beckner, M. & Dailly, A. A pilot study of activated carbon and metal–organic frameworks for methane storage. Appl. Energy 162, 506–514 (2016)
2016
-
[16]
Babaei, H., McGaughey, A. J. H. & Wilmer, C. E. Transient Mass and Thermal Transport during Methane Adsorption into the Metal–Organic Framework HKUST-1. ACS Appl. Mater. Interfaces 10, 2400–2406 (2018)
2018
-
[17]
Huang, B. L. et al. Thermal conductivity of a metal-organic framework (MOF-5): Part II. Measurement. Int. J. Heat Mass Transf. 50, 405–411 (2007). 43
2007
-
[18]
Babaei, H. et al. Observation of reduced thermal conductivity in a metal-organic framework due to the presence of adsorbates. Nat. Commun. 11, 4010 (2020)
2020
-
[19]
& Zhang, X
Huang, J., Fan, A., Xia, X., Li, S. & Zhang, X. In Situ Thermal Conductivity Measurement of Single-Crystal Zeolitic Imidazolate Framework-8 by Raman-Resistance Temperature Detectors Method. ACS Nano 14, 14100–14107 (2020)
2020
-
[20]
Wieser, S. et al. Identifying the Bottleneck for Heat Transport in Metal–Organic Frameworks. Adv. Theory Simul. 4, 2000211 (2021)
2021
-
[21]
A simple nonequilibrium molecular dynamics method for calculating the thermal conductivity
Müller-Plathe, F. A simple nonequilibrium molecular dynamics method for calculating the thermal conductivity. J. Chem. Phys. 106, 6082–6085 (1997)
1997
-
[22]
Li, Z. et al. Influence of thermostatting on nonequilibrium molecular dynamics simulations of heat conduction in solids. J. Chem. Phys. 151, 234105 (2019)
2019
-
[23]
P., Landry, E
Sellan, D. P., Landry, E. S., Turney, J. E., McGaughey, A. J. H. & Amon, C. H. Size effects in molecular dynamics thermal conductivity predictions. Phys. Rev. B 81, 214305 (2010)
2010
-
[24]
Ferreira de Souza, N., Mercier Franco, L. F. & Coasne, B. Consistency of Equilibrium and Nonequilibrium Molecular Dynamics to Assess Thermal Conductivity. J. Chem. Eng. Data 71, 1022– 1032 (2026)
2026
-
[25]
& Cornil, J
Vercouter, A., Lemaur, V., Melis, C. & Cornil, J. Computing the Lattice Thermal Conductivity of Small-Molecule Organic Semiconductors: A Systematic Comparison of Molecular Dynamics Based Methods. Adv. Theory Simul. 6, 2200892 (2023)
2023
-
[26]
Green and Kubo forge the arrow of time
Baroni, S. Green and Kubo forge the arrow of time. Mol. Phys. 123, e2388300 (2025)
2025
-
[27]
& Carbogno, C
Knoop, F., Scheffler, M. & Carbogno, C. Ab initio Green-Kubo simulations of heat transport in solids: Method and implementation. Phys. Rev. B 107, 224304 (2023)
2023
-
[28]
Statistical-Mechanical Theory of Irreversible Processes
Kubo, R. Statistical-Mechanical Theory of Irreversible Processes. I. General Theory and Simple Applications to Magnetic and Conduction Problems. J. Phys. Soc. Jpn. 12, 570–586 (1957)
1957
-
[29]
Green, M. S. Markoff Random Processes and the Statistical Mechanics of Time‐Dependent Phenomena. J. Chem. Phys. 20, 1281–1295 (1952). 44
1952
-
[30]
Ying, P. et al. Sub-Micrometer Phonon Mean Free Paths in Metal–Organic Frameworks Revealed by Machine Learning Molecular Dynamics Simulations. ACS Appl. Mater. Interfaces 15, 36412–36422 (2023)
2023
-
[31]
Wieser, S., Cen, Y.-J., Madsen, G. K. H. & Carrete, J. Accelerating First-Principles Molecular- Dynamics Thermal Conductivity Calculations for Complex Systems. J. Chem. Theory Comput. 22, 513–527 (2026)
2026
-
[32]
Islamov, M. et al. High-throughput screening of hypothetical metal-organic frameworks for thermal conductivity. Npj Comput. Mater. 9, 11 (2023)
2023
-
[33]
Oliveira, L. de S. & Greaney, P. A. Method to manage integration error in the Green-Kubo method. Phys. Rev. E 95, 023308 (2017)
2017
-
[34]
& Baroni, S
Marcolongo, A., Ercole, L. & Baroni, S. Gauge Fixing for Heat-Transport Simulations. J. Chem. Theory Comput. 16, 3352–3362 (2020)
2020
-
[35]
Otero-Lema, M. et al. KUTE: Green–Kubo Uncertainty-Based Transport Coefficient Estimator. J. Chem. Inf. Model. 65, 3477–3487 (2025)
2025
-
[36]
G., Skinner, D
Childers, D. G., Skinner, D. P. & Kemerait, R. C. The cepstrum: A guide to processing. Proc. IEEE 65, 1428–1443 (1977)
1977
-
[37]
& Baroni, S
Ercole, L., Marcolongo, A. & Baroni, S. Accurate thermal conductivities from optimally short molecular dynamics simulations. Sci. Rep. 7, 15835 (2017)
2017
-
[38]
& Baroni, S
Ercole, L., Bertossa, R., Bisacchi, S. & Baroni, S. SporTran: A code to estimate transport coefficients from the cepstral analysis of (multivariate) current time series. Comput. Phys. Commun. 280, 108470 (2022)
2022
-
[39]
& Baroni, S
Bertossa, R., Grasselli, F., Ercole, L. & Baroni, S. Theory and Numerical Simulation of Heat Transport in Multicomponent Systems. Phys. Rev. Lett. 122, 255901 (2019)
2019
-
[40]
& Baroni, S
Pegolo, P., Drigo, E., Grasselli, F. & Baroni, S. Transport coefficients from equilibrium molecular dynamics. J. Chem. Phys. 162, 064111 (2025). 45
2025
-
[41]
S., Gubaev, K., Podryabinkin, E
Novikov, I. S., Gubaev, K., Podryabinkin, E. V. & Shapeev, A. V. The MLIP package: moment tensor potentials with MPI and active learning. Mach. Learn. Sci. Technol. 2, 025002 (2020)
2020
-
[42]
Shapeev, A. V. Moment Tensor Potentials: A Class of Systematically Improvable Interatomic Potentials. Multiscale Model. Simul. 14, 1153–1173 (2016)
2016
-
[43]
& Yaghi, O
Li, H., Eddaoudi, M., O’Keeffe, M. & Yaghi, O. M. Design and synthesis of an exceptionally stable and highly porous metal-organic framework. Nature 402, 276–279 (1999)
1999
-
[44]
S.-Y., Lo, S
Chui, S. S.-Y., Lo, S. M.-F., Charmant, J. P. H., Orpen, A. G. & Williams, I. D. A Chemically Functionalizable Nanoporous Material [Cu3(TMA)2(H2O)3]n. Science 283, 1148–1150 (1999)
1999
-
[45]
Park, K. S. et al. Exceptional chemical and thermal stability of zeolitic imidazolate frameworks. Proc. Natl. Acad. Sci. 103, 10186–10191 (2006)
2006
-
[46]
L., McGaughey, A
Huang, B. L., McGaughey, A. J. H. & Kaviany, M. Thermal conductivity of metal-organic framework 5 (MOF-5): Part I. Molecular dynamics simulations. Int. J. Heat Mass Transf. 50, 393– 404 (2007)
2007
-
[47]
& Liu, L
Zhang, S., Liu, J. & Liu, L. Insights into the thermal conductivity of MOF-5 from first principles. RSC Adv. 11, 36928–36933 (2021)
2021
-
[48]
Effect of pore size and shape on the thermal conductivity of metal-organic frameworks. Chem. Sci. 8, 583–589 (2016)
2016
-
[49]
& Wilmer, C
Islamov, M., Babaei, H. & Wilmer, C. E. Influence of Missing Linker Defects on the Thermal Conductivity of Metal–Organic Framework HKUST-1. ACS Appl. Mater. Interfaces 12, 56172–56177 (2020)
2020
-
[51]
& Jiang, J
Zhang, X. & Jiang, J. Thermal Conductivity of Zeolitic Imidazolate Framework-8: A Molecular Simulation Study. J. Phys. Chem. C 117, 18441–18447 (2013)
2013
-
[52]
& Zhong, Z
Ying, P., Zhang, J., Zhang, X. & Zhong, Z. Impacts of Functional Group Substitution and Pressure on the Thermal Conductivity of ZIF-8. J. Phys. Chem. C 124, 6274–6283 (2020). 46
2020
-
[53]
Lock, N. et al. Elucidating Negative Thermal Expansion in MOF-5. J. Phys. Chem. C 114, 16181–16186 (2010)
2010
-
[54]
Alowasheeir, A. et al. Synthesis of millimeter-scale ZIF-8 single crystals and their reversible crystal structure changes. Sci. Technol. Adv. Mater. 25, 2292485 (2024)
2024
-
[55]
Lee, J. et al. Metal–organic framework materials as catalysts. Chem. Soc. Rev. 38, 1450–1459 (2009)
2009
-
[56]
Pascanu, V., González Miera, G., Inge, A. K. & Martín-Matute, B. Metal–Organic Frameworks as Catalysts for Organic Synthesis: A Critical Perspective. J. Am. Chem. Soc. 141, 7223–7234 (2019)
2019
-
[57]
K., Maddila, S
Gangu, K. K., Maddila, S. & Jonnalagadda, S. B. The pioneering role of metal–organic framework-5 in ever-growing contemporary applications – a review. RSC Adv. 12, 14282–14298 (2022)
2022
-
[58]
Denning, S. et al. Metal–Organic Framework HKUST-1 Promotes Methane Hydrate Formation for Improved Gas Storage Capacity. ACS Appl. Mater. Interfaces 12, 53510–53518 (2020)
2020
-
[59]
Liang, W. et al. Metal–Organic Framework-Based Enzyme Biocomposites. Chem. Rev. 121, 1077–1129 (2021)
2021
-
[60]
Chafiq, M., Chaouiki, A., Suhartono, T. & Ko, Y. G. Albumin protein encapsulation into a ZIF-8 framework with Co-LDH-based hierarchical architectures for robust catalytic reduction. J. Mater. Chem. A 11, 23984–23998 (2023)
2023
-
[61]
Liang, K. et al. Biomimetic mineralization of metal-organic frameworks as protective coatings for biomacromolecules. Nat. Commun. 6, 7240 (2015)
2015
-
[62]
& Hafner, J
Kresse, G. & Hafner, J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B 47, 558– 561 (1993)
1993
-
[63]
& Furthmüller, J
Kresse, G. & Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 6, 15–50 (1996)
1996
-
[64]
& Furthmüller, J
Kresse, G. & Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169–11186 (1996). 47
1996
-
[65]
& Zojer, E
Wieser, S. & Zojer, E. Machine learned force-fields for an Ab-initio quality description of metal-organic frameworks. Npj Comput. Mater. 10, 18 (2024)
2024
-
[66]
& Zojer, E
Strasser, N., Wieser, S. & Zojer, E. Predicting Spin-Dependent Phonon Band Structures of HKUST-1 Using Density Functional Theory and Machine-Learned Interatomic Potentials. Int. J. Mol. Sci. 25, 3023 (2024)
2024
-
[67]
P., Burke, K
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 77, 3865–3868 (1996)
1996
-
[68]
P., Burke, K
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized Gradient Approximation Made Simple [Phys. Rev. Lett. 77, 3865 (1996)]. Phys. Rev. Lett. 78, 1396–1396 (1997)
1996
-
[69]
& Krieg, H
Grimme, S., Antony, J., Ehrlich, S. & Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 132, 154104 (2010)
2010
-
[70]
& Goerigk, L
Grimme, S., Ehrlich, S. & Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 32, 1456–1465 (2011)
2011
-
[71]
& Kresse, G
Jinnouchi, R., Karsai, F. & Kresse, G. On-the-fly machine learning force field generation: Application to melting points. Phys. Rev. B 100, 014105 (2019)
2019
-
[72]
& Kresse, G
Jinnouchi, R., Karsai, F., Verdi, C., Asahi, R. & Kresse, G. Descriptors representing two- and three-body atomic distributions and their effects on the accuracy of machine-learned inter-atomic potentials. J. Chem. Phys. 152, 234102 (2020)
2020
-
[73]
Thompson, A. P. et al. LAMMPS - a flexible simulation tool for particle-based materials modeling at the atomic, meso, and continuum scales. Comput. Phys. Commun. 271, 108171 (2022)
2022
-
[74]
Hamakawa, T., McGaughey, A. J. H. & Shiomi, J. Accurate heat flux formula and thermal conductivity calculation in molecular dynamics simulations with machine learning potentials. J. Appl. Phys. 138, 055105 (2025). 48
2025
-
[75]
T., Wang, C., Cheng, R
Tai, S. T., Wang, C., Cheng, R. & Chen, Y. Revisiting Many-Body Interaction Heat Current and Thermal Conductivity Calculations Using the Moment Tensor Potential/LAMMPS Interface. J. Chem. Theory Comput. 21, 3649–3657 (2025)
2025
-
[76]
F., Knoop, F., Carbogno, C., Scheffler, M
Langer, M. F., Knoop, F., Carbogno, C., Scheffler, M. & Rupp, M. Heat flux for semilocal machine-learning potentials. Phys. Rev. B 108, L100302 (2023)
2023
-
[77]
& Wilmer, C
Boone, P., Babaei, H. & Wilmer, C. E. Heat Flux for Many-Body Interactions: Corrections to LAMMPS. J. Chem. Theory Comput. 15, 5579–5587 (2019)
2019
-
[78]
The fluctuation-dissipation theorem
Kubo, R. The fluctuation-dissipation theorem. Rep. Prog. Phys. 29, 255 (1966)
1966
-
[79]
Hardy, R. J. Energy-Flux Operator for a Lattice. Phys. Rev. 132, 168–177 (1963)
1963
-
[80]
& Maiti, P
Mandal, S. & Maiti, P. K. Prediction of thermal conductivity in CALF-20 with first-principles accuracy via machine learning interatomic potentials. Commun. Mater. 6, 22 (2025)
2025
-
[81]
Howell, P. C. Comparison of molecular dynamics methods and interatomic potentials for calculating the thermal conductivity of silicon. J. Chem. Phys. 137, 224111 (2012)
2012
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.