Effective Biological Representation Learning by Masking Gene Expression
Pith reviewed 2026-06-28 23:10 UTC · model grok-4.3
The pith
A masked autoencoder on a curated 1.4 million RNA-seq samples produces higher-fidelity gene representations than foundation models trained on over 100 times more data.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
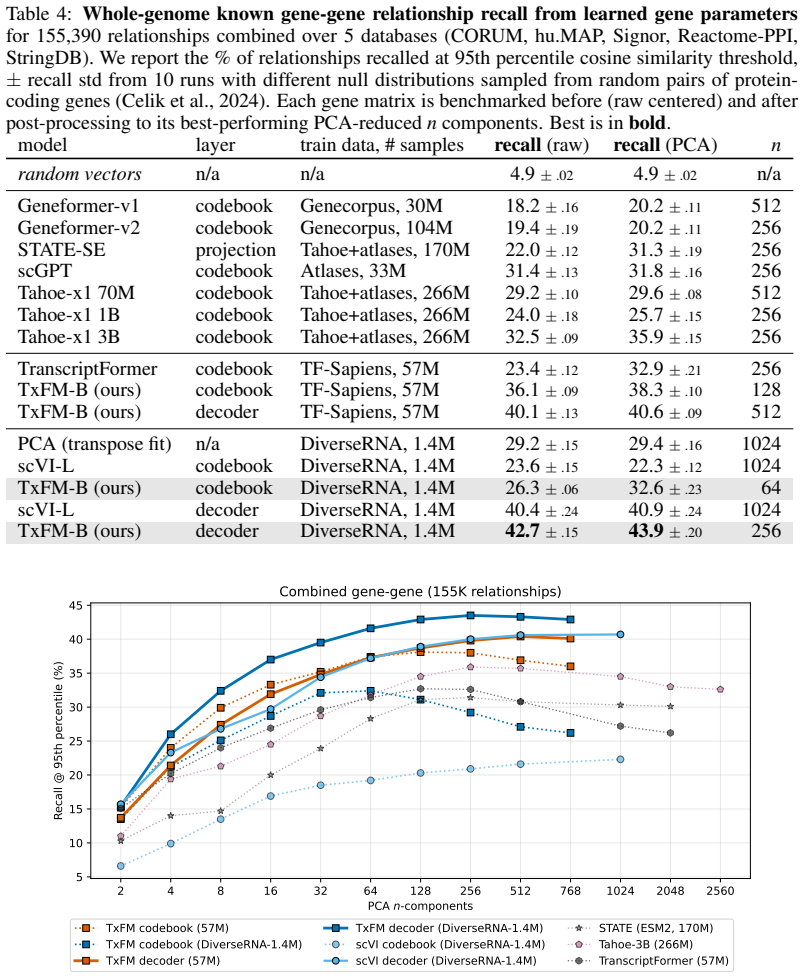
TxFM employs a masked autoencoding approach tailored to diverse RNA-seq count data; when trained on the curated DiverseRNA-1.4M corpus, it yields high-fidelity gene representations that outperform foundation models trained on atlas-scale corpora over 100x larger, indicating that inductive self-supervised learning is viable for transcriptomics representation provided careful synthesis of architecture and training data curation.
What carries the argument
Masked autoencoding tailored to RNA-seq count data, with architecture ablations that identify configurations required for strong transfer.
If this is right
- Inductive self-supervised learning becomes a practical route for transcriptomic representation learning.
- Data curation can matter more than raw corpus scale for building effective gene embeddings.
- Architecture choices identified in the ablations become necessary components for strong transfer in similar models.
- Raw transcript counts are no longer the default baseline once masked pretraining is applied correctly.
Where Pith is reading between the lines
- Drug-discovery pipelines that rely on gene-expression signatures could substitute TxFM embeddings for raw counts or larger-model outputs without increasing compute.
- The same masking strategy might extend to other count-based biological modalities if the count-distribution handling is preserved.
- Future work could test whether adding explicit batch-effect correction inside the masking objective further improves out-of-distribution transfer.
Load-bearing premise
The inductive evaluations compare models fairly without data leakage or post-hoc selection, and the curated DiverseRNA-1.4M dataset represents the distribution of target downstream tasks.
What would settle it
A downstream gene-expression task in which TxFM embeddings produce lower performance than a linear baseline or a model trained on a larger atlas after controlling for evaluation protocol and data overlap.
Figures
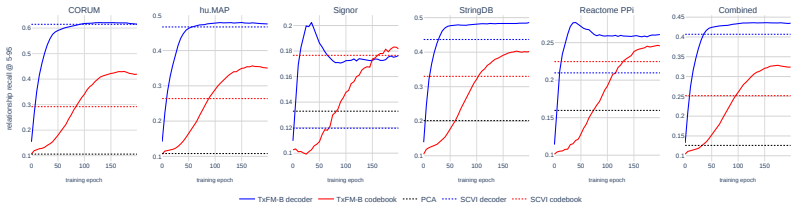
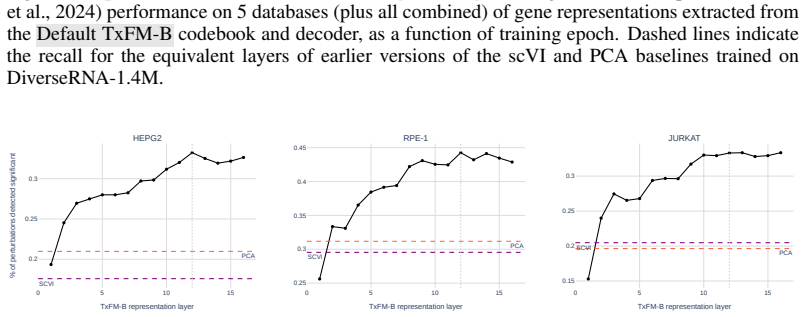
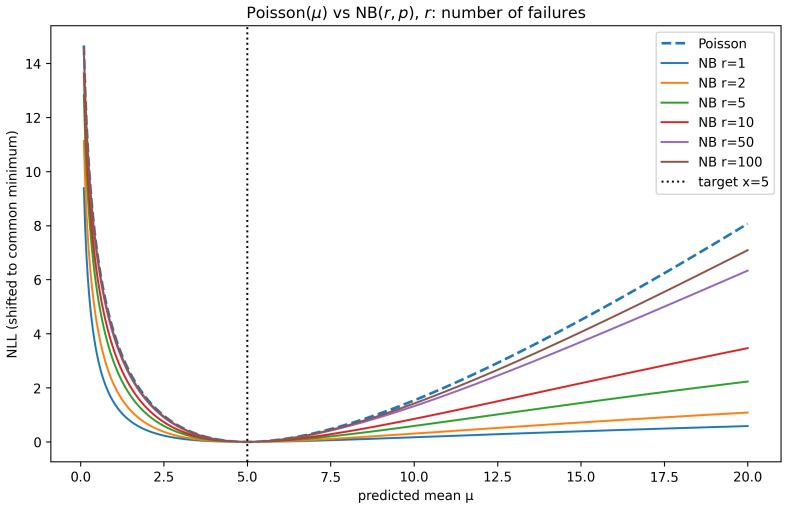
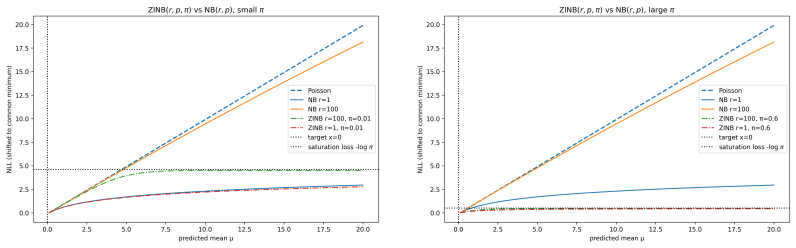
read the original abstract
RNA sequencing produces rich and diverse datasets of gene expression, offering compelling insights into cellular state and function that have many applications in drug discovery. Modeling such data is challenging due to inherent technical noise and experimental batch effects, as evidenced by many existing transcriptomic foundation models (FMs) underperforming relative to linear baselines. Such results raise the question of whether deep representation learning provides a distinct advantage over the direct use of raw transcript counts. Our work explores this by developing a new self-supervised model, TxFM, with a focus on inductive representation learning evaluations. TxFM employs a masked autoencoding approach tailored to diverse RNA-seq count data, and our ablation study empirically identifies crucial architecture configurations required for strong transfer performance. Additionally, we curate a public training corpus, DiverseRNA-1.4M, and find that TxFM trained on this curated dataset yields high-fidelity gene representations that outperform FMs trained on atlas-scale corpora over 100x larger. Overall, our results indicate that inductive self-supervised learning is a viable modeling approach for transcriptomics representation, provided a careful synthesis of model architecture and training data curation.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript introduces TxFM, a masked autoencoder for self-supervised learning of gene representations from RNA-seq count data. It curates the DiverseRNA-1.4M training corpus and reports that TxFM trained on this dataset produces representations that outperform transcriptomic foundation models trained on atlas-scale corpora more than 100 times larger, based on inductive representation learning evaluations; an architecture ablation is also presented to identify key design choices for transfer performance.
Significance. If the outperformance claim holds under strictly inductive conditions with consistent evaluation protocols, the result would demonstrate that targeted data curation combined with appropriate architecture choices can yield stronger biological representations than scale alone, providing a counterpoint to the prevailing emphasis on ever-larger training corpora in transcriptomics foundation modeling.
major comments (1)
- [Abstract] Abstract: the central claim that TxFM on DiverseRNA-1.4M outperforms FMs trained on >100x larger corpora is load-bearing for the paper's contribution, yet the manuscript supplies no quantitative verification that downstream task splits have zero overlap with DiverseRNA-1.4M, that identical task definitions/metrics/splits were applied to all baselines, and that no post-hoc model or hyperparameter selection occurred after inspecting test results.
Simulated Author's Rebuttal
We thank the referee for their thorough review and for highlighting the importance of rigorous verification for our central inductive transfer claims. We address the major comment below and commit to revisions that will strengthen the manuscript's transparency and reproducibility.
read point-by-point responses
-
Referee: [Abstract] Abstract: the central claim that TxFM on DiverseRNA-1.4M outperforms FMs trained on >100x larger corpora is load-bearing for the paper's contribution, yet the manuscript supplies no quantitative verification that downstream task splits have zero overlap with DiverseRNA-1.4M, that identical task definitions/metrics/splits were applied to all baselines, and that no post-hoc model or hyperparameter selection occurred after inspecting test results.
Authors: We agree that explicit verification of these points is essential to substantiate the load-bearing claim of outperformance under strictly inductive conditions. The current manuscript does not include quantitative overlap checks or a consolidated description of protocol consistency across models. In the revised version, we will add a new appendix (or expanded methods section) that: (1) provides quantitative verification of zero overlap between downstream task splits and DiverseRNA-1.4M via explicit checks on sample and gene identifiers; (2) documents the precise task definitions, metrics, and data splits used for every baseline, confirming they were applied identically; and (3) details the hyperparameter selection workflow, which relied exclusively on validation performance with no access to test results at any stage. These additions will directly address the referee's concerns while preserving the paper's focus on data curation and architecture choices. revision: yes
Circularity Check
No derivation chain present; claim is purely empirical
full rationale
The paper's central claim is an empirical performance comparison: TxFM trained on DiverseRNA-1.4M outperforms larger atlas-scale FMs on inductive downstream tasks. The abstract and description contain no equations, derivations, fitted parameters renamed as predictions, or self-referential definitions. No load-bearing steps reduce to inputs by construction. The evaluation protocol concerns (inductive splits, baseline consistency) are questions of experimental validity, not circularity in a derivation. Self-contained empirical result with no visible reduction.
Axiom & Free-Parameter Ledger
Reference graph
Works this paper leans on
-
[1]
doi: 10.1101/2025.06.26.661135. URLhttps://www.biorxiv.org/content/ early/2025/07/10/2025.06.26.661135. Constantin Ahlmann-Eltze, Wolfgang Huber, and Simon Anders. Deep-learning-based gene per- turbation effect prediction does not yet outperform simple linear baselines.Nature Methods, 22: 1657–1661, 2025. doi: 10.1038/s41592-025-02772-6. URLhttps://doi.or...
-
[2]
the proportion of the variance that is explained by the batch variable between the original dataset and the embeddings of the model
counts pairs that are consistently assigned (same cluster in both clusterings: nS; different clusters in both:n D).ARIis more conservative thanNMI, requiring precise boundary correspondence for high scores. Average Silhouette Width (ASW):Measures separation of true cell types in embedding space: ASW = 1 n nX i=1 b(l) i −a (l) i max(a(l) i , b(l) i ) (22) ...
2025
-
[3]
Removing all K562 cells from the training data, yielding 932K training samples
-
[4]
(2022) dataset, in addition to the 932K non-K562 samples in the DiverseRNA dataset
Adding only the 72,000 non-targeting control K562 cells from the Replogle et al. (2022) dataset, in addition to the 932K non-K562 samples in the DiverseRNA dataset
2022
-
[5]
Our default approach, which follows an established strategy shown to improve the perfor- mance of MAEs on biological experimental data (Kenyon-Dean et al., 2025) by filtering the perturbational K562 training data to only include cells from perturbations distinguishable from the rest. Specifically, we selectedphenoprintperturbations by using an earlier ver...
2025
-
[6]
Training with the full uncurated dataset of K562 cells, yielding 2.8M training samples, and holding the compute budget constant by adjusting the number of training epochs according to dataset size; we also evaluate doubling the training compute on the 2.8M dataset to 200 epochs [2x], to about 2,000 H100 GPU hours
-
[7]
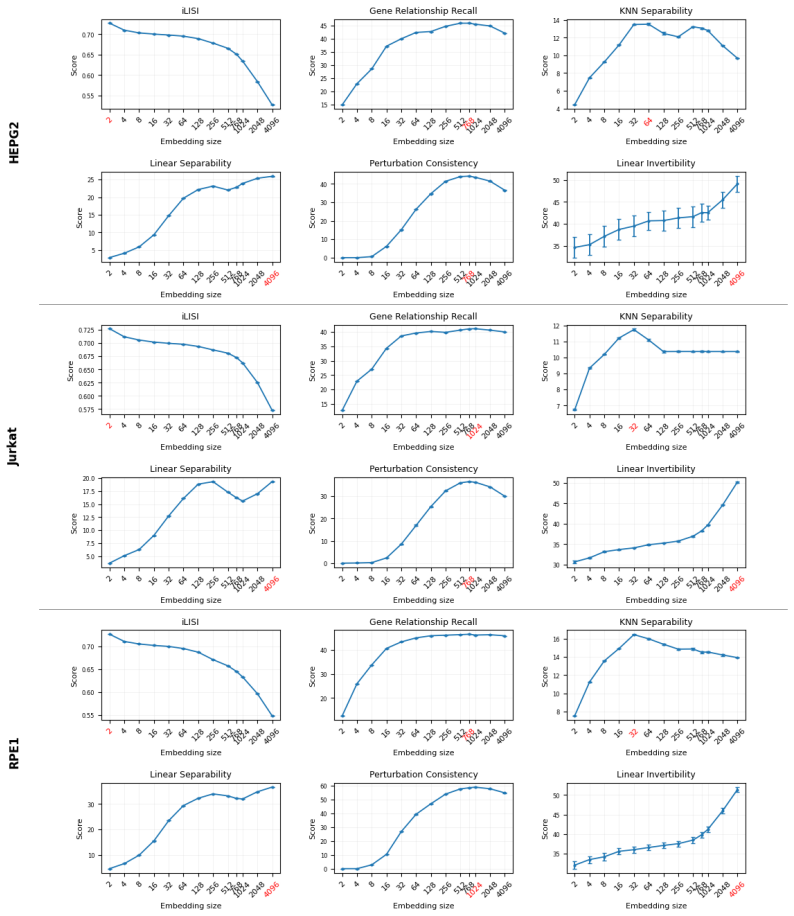
Training TxFM-B with 4x more compute on a different dataset of 57 million cells sampled from 72 large-scale atlases from CZI et al. (2025). 28 Table 9:Backbone choice for existing models. Average score across all benchmarking tasks in Bendidi et al. (2024) on RPE1, HEPG2, and Jurkat datasets for different backbone choices of exist- ing FMs with multiple p...
2025
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.